中温煤焦油沥青逐级萃取的产物组成与结构分析
2020-12-17崔瀛丹哈丽丹买买提
崔瀛丹,钟 梅,哈丽丹·买买提,杨 超,樊 星
(1.新疆大学 化工学院,新疆 乌鲁木齐 830046; 2.新疆大学 新疆煤炭清洁转化与化工过程新疆维吾尔自治区重点实验室,新疆 乌鲁木齐 830046)
煤沥青是煤焦油蒸馏提取轻质组分后的残留物,约占焦油的50%~60%,其基本结构单元是稠环芳烃及其衍生物[1-2]。高温煤沥青主要用于生产沥青焦和活性炭,是制备碳纤维[3-5]、碳量子点[6-8]、碳基复合材料[9-11]等高性能碳材料的优良前驱体。中温煤沥青软化点为75~95 ℃,具有含碳量高、流动性好和易石墨化等优点,主要用于生产炭材料、粘结剂和浸渍剂[12-13](如生产石墨电极、电解铝用碳槽、阳极糊和镁碳砖等)。低温沥青中富含长链烷烃,具有润湿和粘附能力强、耐油侵蚀、行车安全系数大、路面摩擦因数大等优势,目前主要用作筑路沥青[14-15]。
由于煤沥青组分十分复杂,杂原子含量高且含有较多的有害金属杂质,在催化剂表面易于沉积,造成催化剂孔道堵塞导致催化剂失活,因此研究其组成与结构十分必要[16]。借助现代仪器的分析测试,研究者们给出了较为详细的结构信息[17-18]。庞伟伟[19]采用元素分析、GCP、NMP和FITR对煤沥青进行了表征,使用改进的Brown-Ladner结构参数计算模型构建了以稠环芳烃为主体的煤沥青平均分子结构模型。高丽娟等[20]分析了改性沥青的结构,结合Brown-Lander参数模型计算得到其分子结构的平均芳环数为8,芳烃分子以面性排列为主。
依据“相似相溶”原理,对煤沥青进行族组分分离,有利于获取更为详尽的结构信息。E.J狄金松[21]使用吡啶、苯和正己烷将煤沥青分为油分、树脂A、树脂B、吡啶不溶物和吡啶可溶甲苯不溶5个组分。GILBERT MORGAN[22]采用脂烃类溶剂(正己烷或轻石油馏分)对煤沥青进行溶解,将其可溶物命名为结晶质。MCNEIL和WOOD等[23]使用轻石油、苯、硝基苯和喹啉将煤沥青分成5个组分,其中结晶质A和B可溶于轻石油,收率为54.6%;树脂可溶于苯但不溶于轻石油,收率为17.3%;溶于硝基苯但不溶苯的组分收率为15.4%;溶于喹啉但不溶硝基苯组分收率为6.5%,总收率达93.8%,通过测定5种组分的分子量分布,发现50%的组分分子量分布在150~300 u,其余产物分子量分布在1 400~5 000 u。
笔者以中温煤焦油沥青为原料,通过Hansen三维溶解度参数软件(HSPIP)[24]选择适宜的萃取剂对煤沥青进行逐级分离,采用高分辨率质谱、X射线光电子能谱(XPS)、固体超导核磁共振(13C NMR)、傅里叶变换红外光谱(FTIR)、X射线衍射(XRD)和热重分析仪(TG)等表征分析各级萃取物和萃余物,获悉中温煤沥青的组成、元素的相对含量及禀赋形态、碳骨架信息、官能团特征和共价键类型等,以期为煤沥青的深加工利用提供数据支撑和理论依据[25]。
1 实验部分
1.1 原 料
中温煤焦油沥青取自榆林(RM),首先对其进行破碎、筛分,选取粒径<200目的中温沥青于50 ℃的真空干燥箱中干燥12 h后,放置自封袋中密封备用,其元素分析见表1。

表1 样品元素分析Table 1 Ultimate analysis of sample %
1.2 实验过程
1.2.1萃取剂选取
通过(HSPIP)模拟确定各级萃取剂[26]。该软件采用溶剂对材料的溶胀效果来模拟计算其三维溶解度参数,即分散力(δd)、极性力(δp)和氢键力(δh)。实验所用溶剂共36种,均为分析纯(北京北化精细化学品有限公司),包括正戊烷(n-C5)、正己烷(n-C6)、正庚烷(n-C7)、正辛烷(n-C8)、正壬烷(n-C9)、正癸烷(n-C10)、十一烷(n-C11)、十二烷(n-C12)、苯、甲苯、对二甲苯、乙苯、二氯甲烷、三氯甲烷、四氯化碳、四氢呋喃、1,4-二氧六环、环己烯、辛烯、丙酮、戊酮、环己酮、乙醚、丁醚、甲酸甲酯、乙酸甲酯、乙酸乙酯、丙酸甲酯、乙醇、丙醇、异丙醇、丁醇、异丁醇、乙腈、N,N-二甲基甲酰胺和N-甲基吡咯烷酮。
1.2.2逐级萃取
称取30 g左右中温煤沥青置于滤纸桶中,放入索氏抽提器中选用约300 mL的无水乙醇萃取至无色,利用旋转蒸发仪对萃取剂进行分离,将分离后的萃余物真空干燥至质量基本不发生变化,然后选用下一级萃取剂继续此过程,直至完成六级萃取。其流程如图1所示。
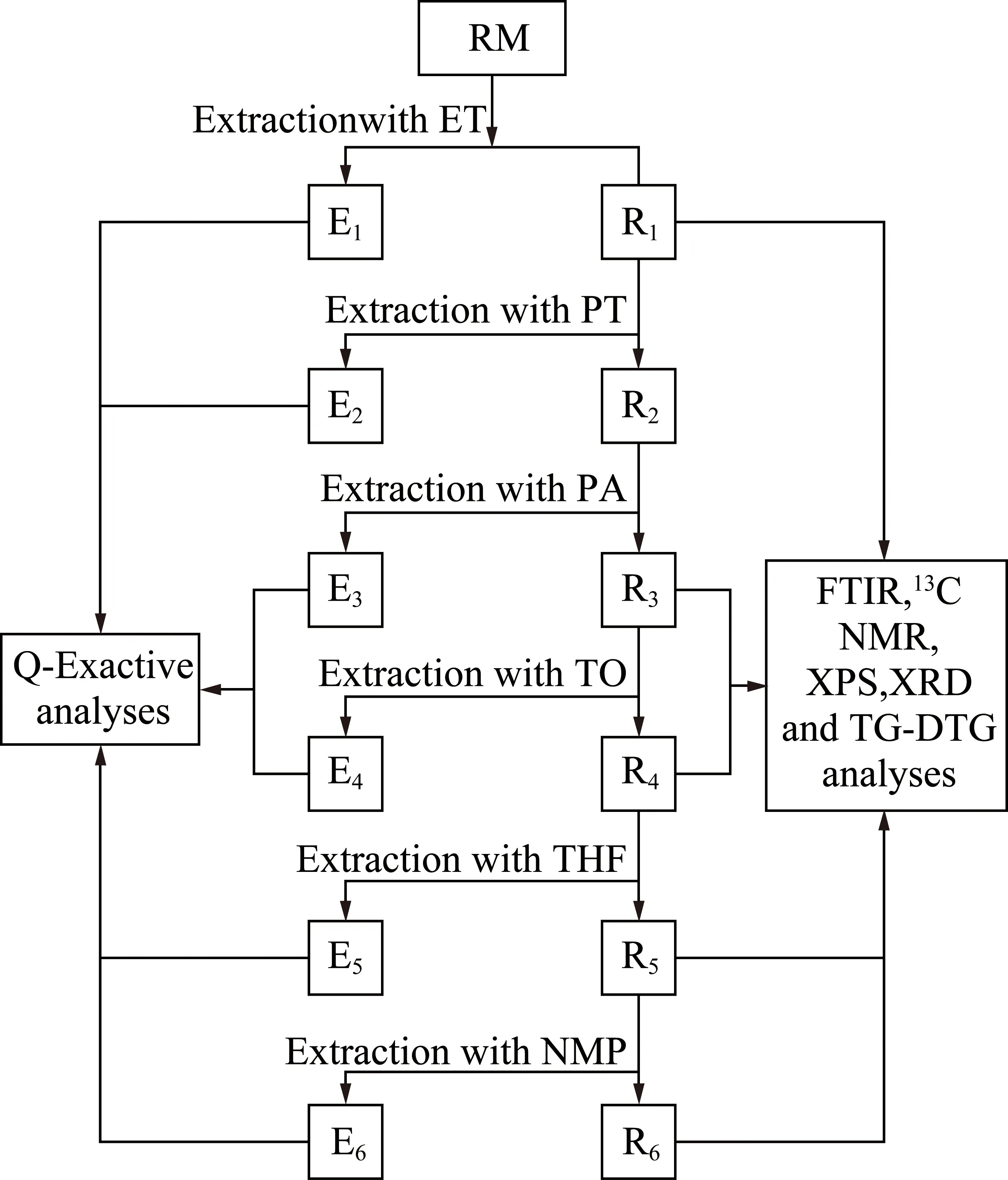
图1 煤沥青的逐级索氏萃取流程Fig.1 Sequential soxhlet extraction of coal tar pitch
1.3 分析测试方法
采用美国TA公司生产的SDT-Q600热重分析仪测定样品的热失重行为:样品质量约5 mg,以100 mL/min的高纯氩气为载气,升温速率为10.0 ℃/min,终温1 000 ℃。
试样的分子量分布由美国Thermo Fisher Scientific 公司生产的轨道阱质谱(Q-Exactive)分析,离子源类型为大气压化学电离源(APCI)。主要仪器参数:正离子APCI电离模式,发射电压 5 000 V,毛细管入口电压 5 500 V,毛细管出口电压 320 V。采集质量150~1 000 Da。
在日本Rigaku D/max2500型X射线衍射仪上测定样品的晶相结构:Cu靶Kα射线(λ=0.154 056 nm),Ni滤波,扫描速率8°/min,石墨单色管,管电压和管电流分别为40 kV和100 mA,步长0.01°,扫描范围5°~85°。
样品的官能团及骨架信息由德国Bruker公司生产的EQUINOX-55傅里叶变换红外光谱仪测定,分辨率为0.4 cm-1,扫描区间为500~4 000 cm-1,波数精度0.01 cm-1,累加扫描次数16次。样品∶溴化钾质量比=1∶400。
碳类型分布由美国Varian公司生产的Varian Ino va-400超导核磁共振谱仪测定:固体双共振探头,转速为5 kHz,共振频率为100.38 MHz,循环延迟时间6 s,碳氢交叉极化接触时间2 ms,数据扫描采集共计9 000次。
采用美国Thermo Fisher Scientific 公司生产的ESCALAB 250Xi X射线光电子能谱仪分析样品的元素存在形态,单色化AI K AIpha 射线源,功率150 W,以C1s(284.6 eV)为标准进行能量校正。
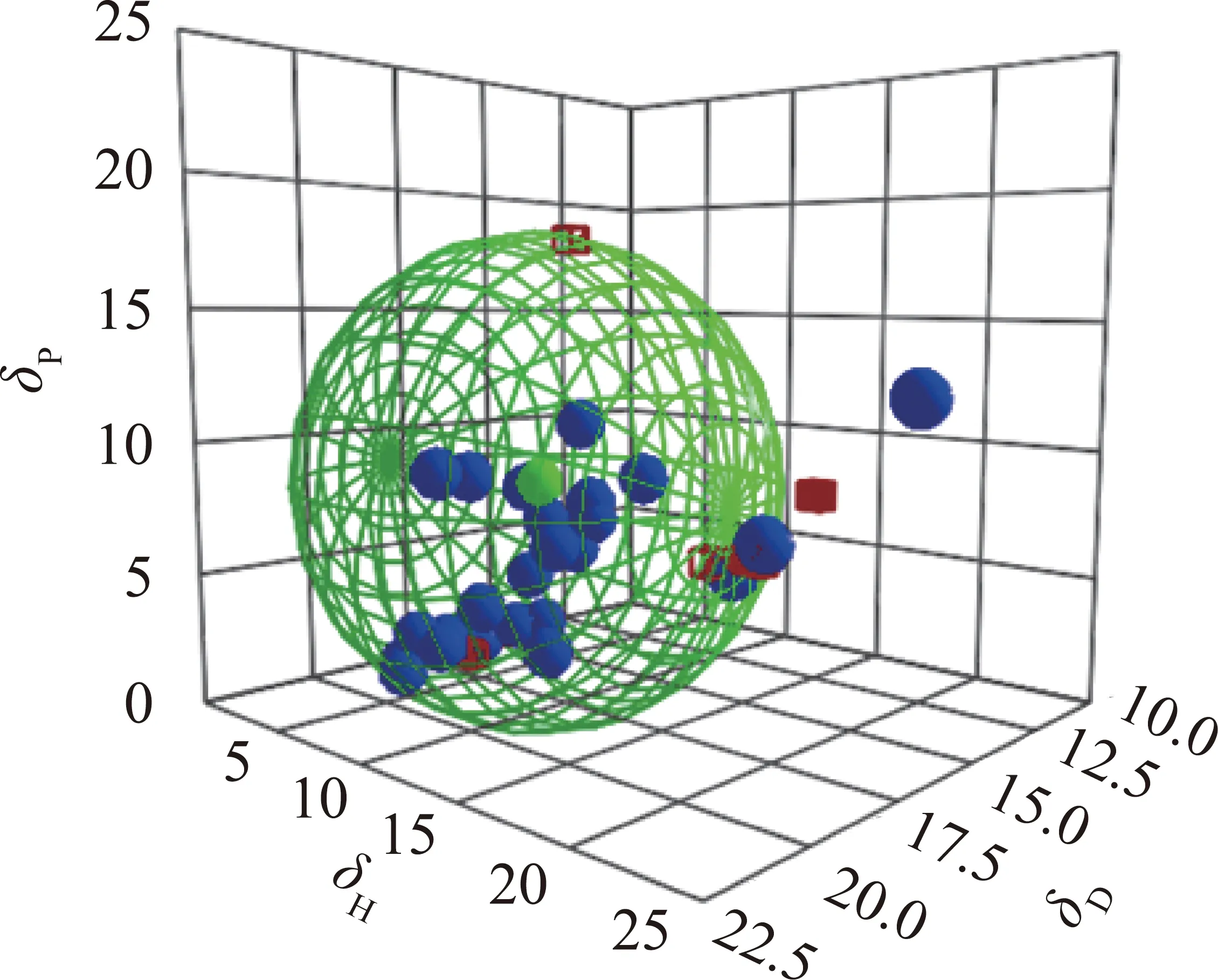
图2 Hansen 三维溶解度球示意Fig.2 Hansen solubility sphere in a 3D diagram
2 结果与讨论
2.1 萃取剂选择
原料的 “Hansen 三维溶解度球”如图2所示,各溶剂的汉森三维溶解度参数(HSPs)值和相应的溶胀度见表2。图中绿色小球表示样品,蓝色小球代表良溶剂,位于溶解度参数球内;红色正方体则代表不良溶剂,位于参数球外。将模拟计算优化后的“Hansen 三维溶解度球”半径记为R0,各有机溶剂与样品在球中的距离记为Ra,相对能量差RED=Ra/R0。参考相关文献[27],萃取剂应选取溶解度参数球边沿且RED值接近1的蓝色小球较为合适。故分别选取ET,PT,PA,TO,THF,NMP作为1~6级萃取剂,相应的萃取物分别记作ETE,PTE,PAE,TOE,THE,NME,萃余物分别记作ETR,PTR,PAR,TOR,THR,NMP萃余物残渣仅为3.29%,所以只对1~5级萃余物进行表征分析。
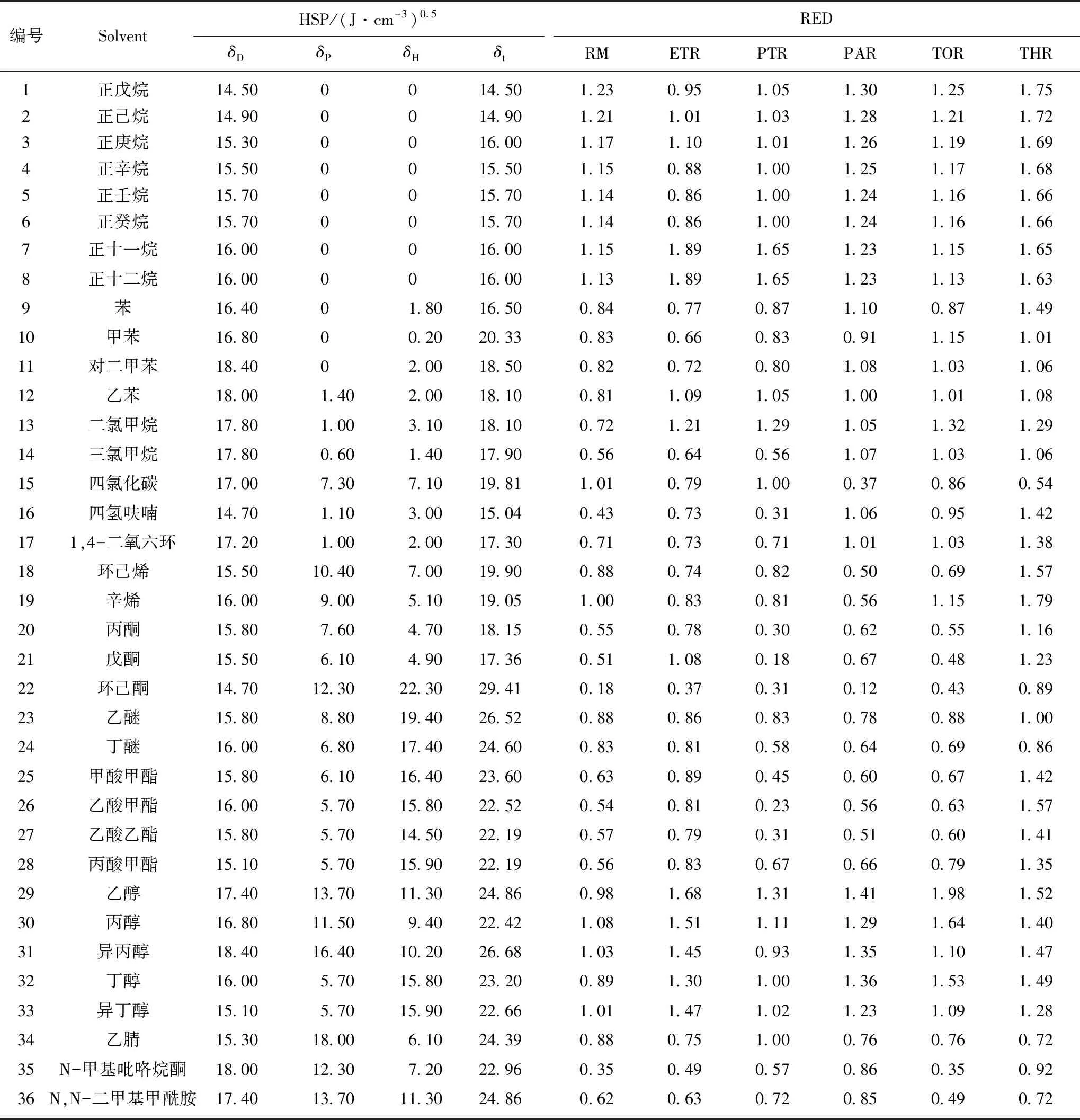
表2 溶剂的HSPs值和相应的溶胀度Table 2 HSPs of solvents and the measured swelling degree
2.2 萃取物与萃余物结构分析
经无水乙醇(ET)、正戊烷(PT)、异丙醇(PA)、甲苯(TO)、四氢呋喃(THF)和N-甲基吡咯烷酮(NMP)萃取后,各级萃取率分别为0.69%,5.64%,11.57%,32.78%,11.52%和34.51%,残渣仅为3.29%。
2.2.1萃取物分析
萃取物的高分辨质谱结果如图3所示。从图3可以看出,各级萃取物的分子量分布范围为100~400 u,鉴定出的化合物多达3 000多种,其中相对含量较高的20种化合物中杂环类为9种,见表3。采用通用分子式CcHhOoNnSs计算不饱和度的双键当量(DBE),DBE=c-(h-n)/2+1[28]。根据杂原子分布,将含有杂原子的化合物分为Oo,Nn,Ss,ONS(包含OoNn,OoSs,NnSs,OoNnSs)4类[29],不含杂原子的化合物记为CH类化合物,DBE及碳数分布和各类化合物相对含量分布结果如图4所示。
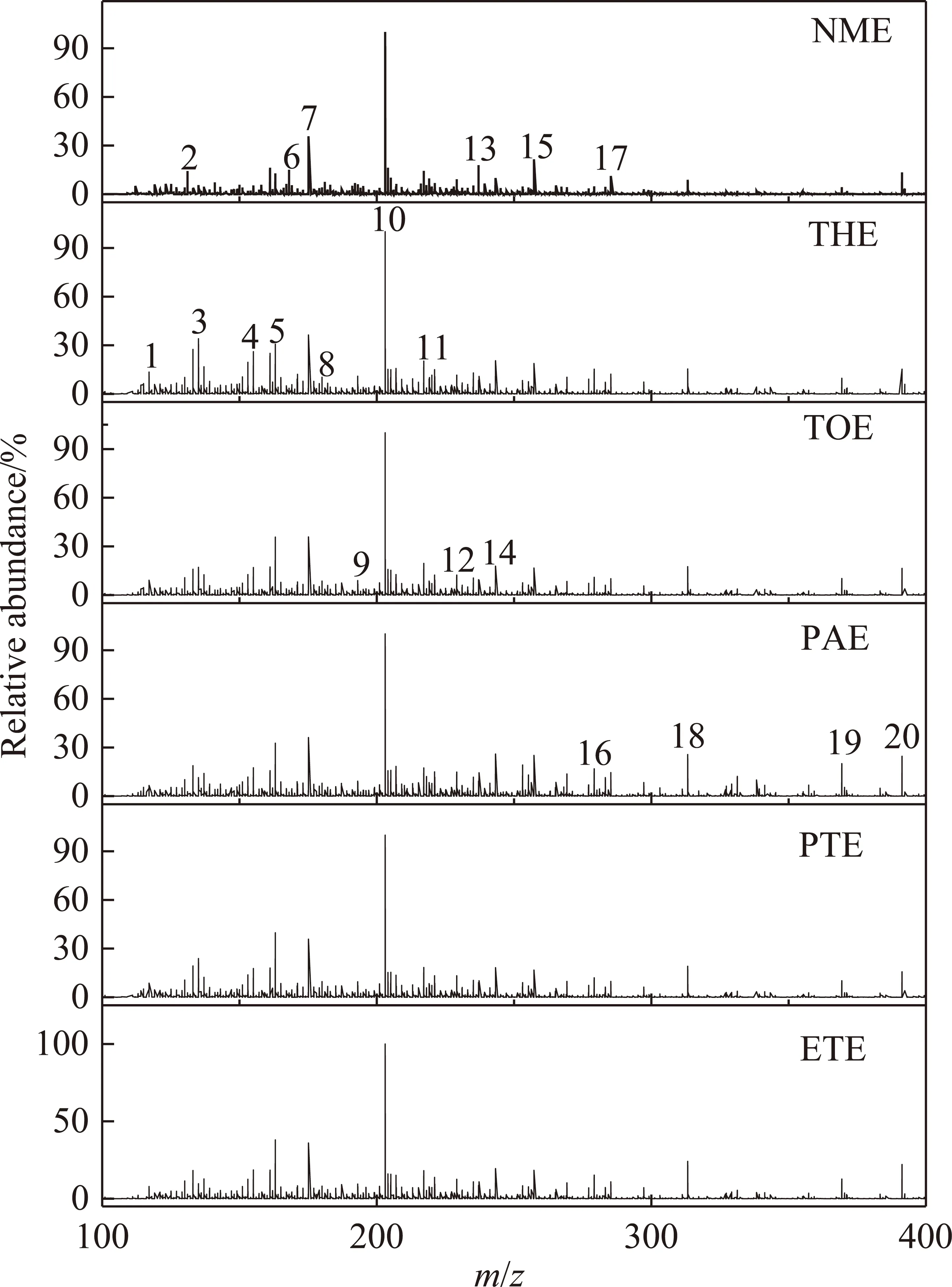
图3 萃取物的轨道阱质谱图Fig.3 Mass spectra of extracts obtained by orbitrap-MS

表3 萃取物中主要化合物Table 3 Main compounds in extracts
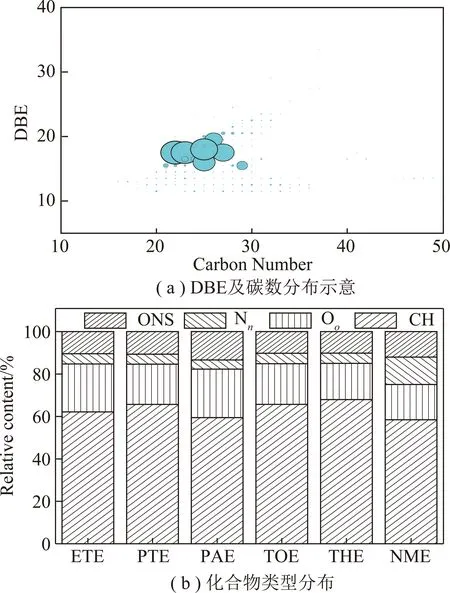
图4 样品的轨道阱质谱分析Fig.4 Mass spectra of samples obtained by orbitrap-MS
图4(a)为ETE的DBE及分布图,其余几级萃取物与之类似,从图4(a)可以看出,萃取物的DBE值主要分布在15~19,碳数为21~28。文献表明[30],对于芳烃化合物,无烷基取代的苯并芘、苝、苯并荧蒽分子的DBE=15,碳原子数为20;无烷基取代的二苯并芘、苯并苝分子的DBE=18,碳原子数为24;有1~3个烷基取代的5环芳烃化合物的DBE值为15~17,碳数为21~24;有1~2个烷基取代的6环芳烃化合物的DBE值在18~19,碳数则为24~28。故推测各级萃取物以含有1~3个烷基取代的5~6环芳烃化合物为主。
图4(b)给出了各级萃取物中5类化合物的含量分布。由图4(b)可知各级萃取物中含量较高的均为CH类化合物,比例高达60%,杂原子化合物中Oo类的相对含量较高,ETE和PAE中Oo类化合物含量高于其他几级萃取物,这是因为醇类溶剂极性较大,可以与沥青中的有机小分子形成氢键,削弱了沥青中原有氢键,从而萃取出较多的含氧类化合物[31]。Nn类化合物相对含量为4.3%~12.9%,主要是吡啶型和吡咯型氮化物,从图4(b)可以看出NME中Nn类化合物明显高于其它几级萃取物,这是因为NMP中吡咯烷酮环状结构可以与沥青中芳香结构相互作用,破环沥青中小分子间的相互作用力与之结合,因此NME中含氮化合物较高[32]。Ss类化合物未被检测出,这是因为此类化合物极性很低,相对来说比较难电离,而分子中存在氧原子和氮原子时,极性增加[33],因此其他类含硫化合物(OoSs,NnSs和OoNnSs)可被检测到,ONS类化合物的相对含量之和为10.1%~12.0%。
2.2.2萃余物结构分析
(1)热重分析。从各级萃余物的TG和DTG图中可以看出(图5),除TOR和THR外,RM,ETR,PTR和PAR的热分解曲线相似,说明4者的热解过程较为接近,对其进行温度区间划分:第1段为室温~220 ℃,各物质主要发生软化、熔融、微量水分脱除、部分弱共价键断裂、轻质组分少量逸出并伴随轻微的热分解反应,RM,ETR,PTR和PAR的失重量分别为5.04%,5.07%,6.28%和5.77%;第2阶段为220~480 ℃,是主要热解段,挥发物大量逸出,在350 ℃左右,各物质失重速率达到最大值,归属于Cal—Cal,Cal—H等较强共价键的断裂,在435 ℃左右出现失重速率极值峰,归属于Car—Cal,Car—O,Car—S等强共价键断裂[34],所涉及的反应包括:① 热分解/聚合反应产生的小分子和气态产物的去除;② 较高温度下含氧杂环化合物的分解[35]。该段RM,ETR,PTR和PAR的失重量分别为54.4%,55.37%,53.74%和49.49%,即随萃取级数增加,萃余物的失重量减少,且失重速率极值峰温右移,这是由于随着萃取级数增加,各萃取物的分子量增加,不易分解所致。第3阶段为480~800 ℃,RM,ETR,PTR和PAR于510 ℃出现失重速率极值峰,可归因于稠环芳烃化合物缩聚过程中脱出的烷基和氢等小分子物质[36]。
甲苯萃余物TOR和四氢呋喃萃余物THR的总失重量仅分别为19.79%和18.52%,主要失重阶段为220~600 ℃,且峰温右移至510 ℃,说明TOR和THR中高分子量的组分多,侧链等基团含量少,因而热稳定增大。
(2)FTIR分析。从图6可以看出,原料及各级萃余物均在波数3 442 cm-1处出现—OH伸缩振动吸收峰,说明各级萃取剂能将沥青中的部分酚类化合物溶出。波数3 040 cm-1和1 616 cm-1处为芳环上C—H和C=C骨架伸缩振动吸收峰。可以看出,随着萃取级数的增加,3 040 cm-1的芳香C—H强度减弱,说明芳环缩合度逐渐增加。PAR和THR在1 616 cm-1处的C=C骨架伸缩振动吸收峰明显增强。波数2 960和1 427 cm-1处出现脂肪链—CH3的伸缩振动和弯曲振动峰,说明原料及各级萃余物中均存在脂肪侧链;波数1 043 cm-1处为C—O的伸缩振动峰,THR的峰强度明显高于其它物质,说明其大分子结构中含有较多的醚键;波数在880~680 cm-1内出现了874,812和746 cm-1三种类型的苯环上的C—H面外弯曲振动特征峰。
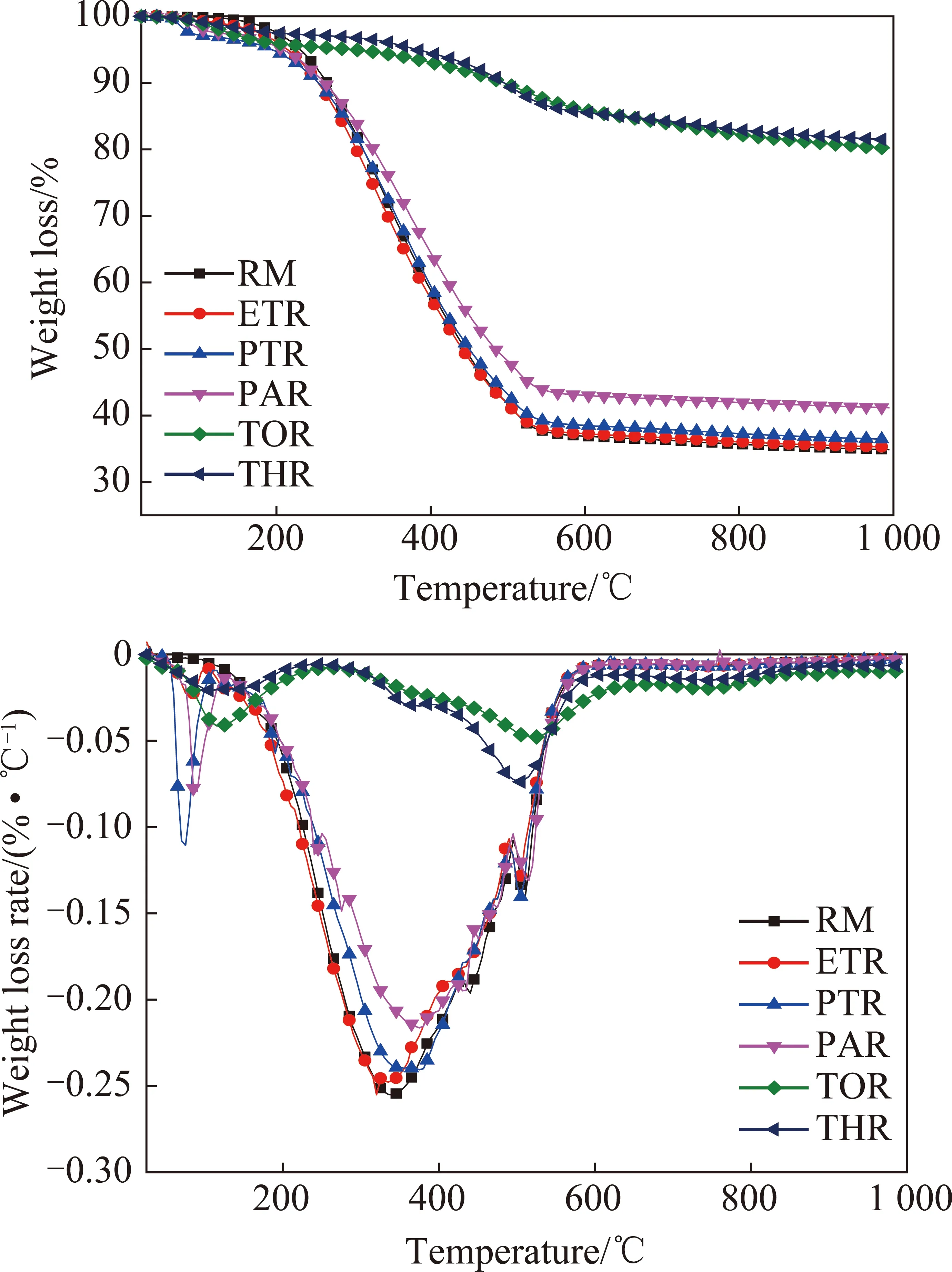
图5 萃余物的TG-DTG分析Fig.5 TG-DTG analysis of residues
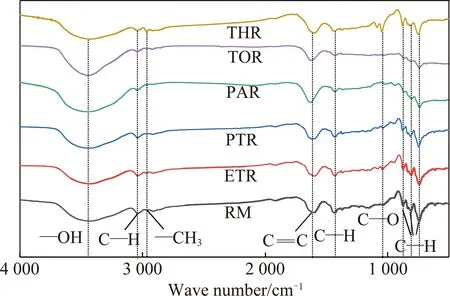
图6 原料及萃余物的红外分析Fig.6 FTIR analysis of raw pitch and residues
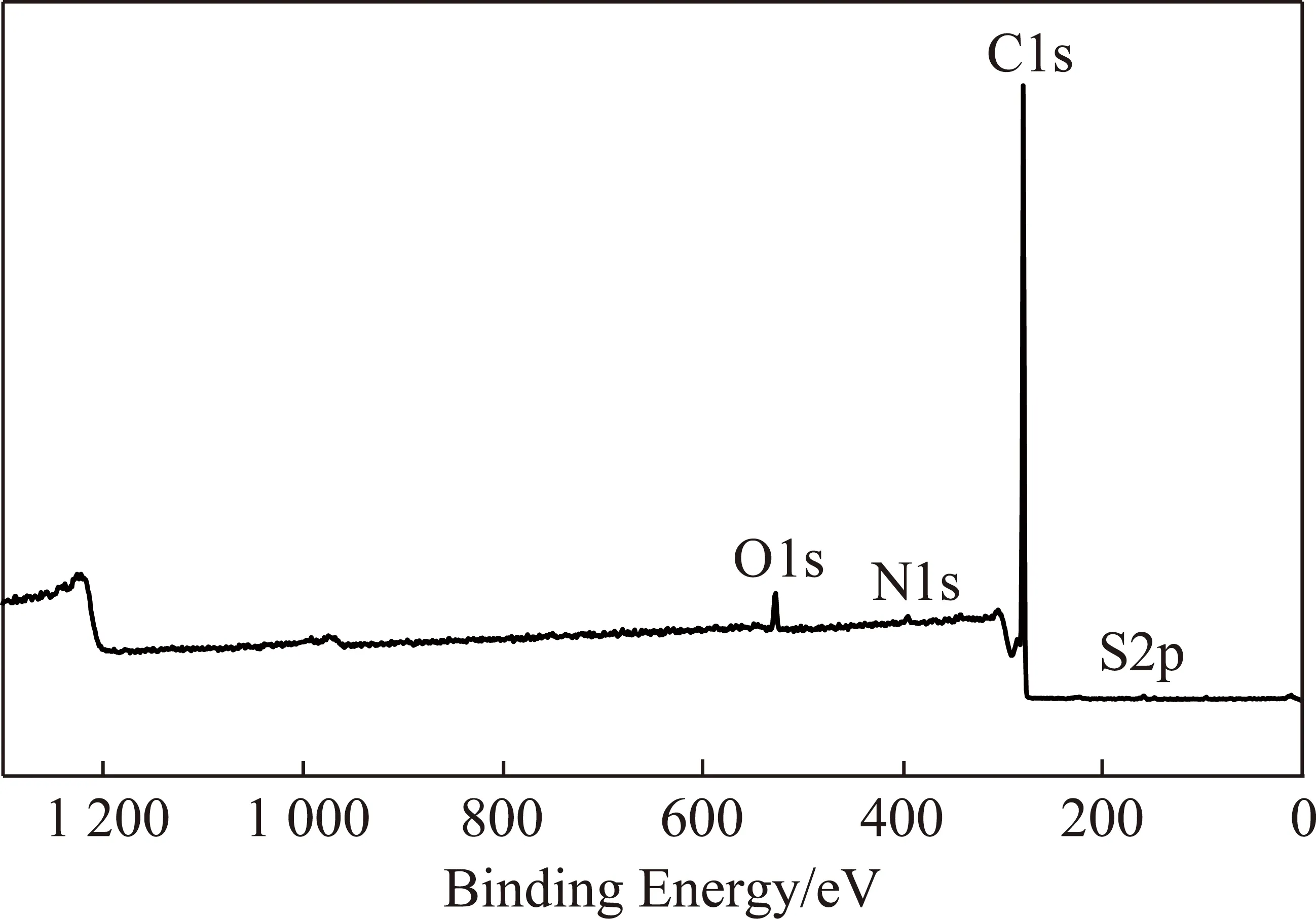
图7 沥青的XPS谱图Fig.7 XPS spectrum of pitch
(3)元素形态分析。图7给出了沥青及各级萃取物的XPS谱图。从图7可以看出,沥青表面的主要元素是C和O,N和S含量很少。为了分析沥青及各级萃余物表面C,O,N和S的赋存形态及相对含量,分别对其进行窄区扫描,用Peak Fit对各峰进行分峰拟合,结果如图8和表4。

图8 原料的XPS分峰拟合曲线Fig.8 Fitting curves of XPS for raw material
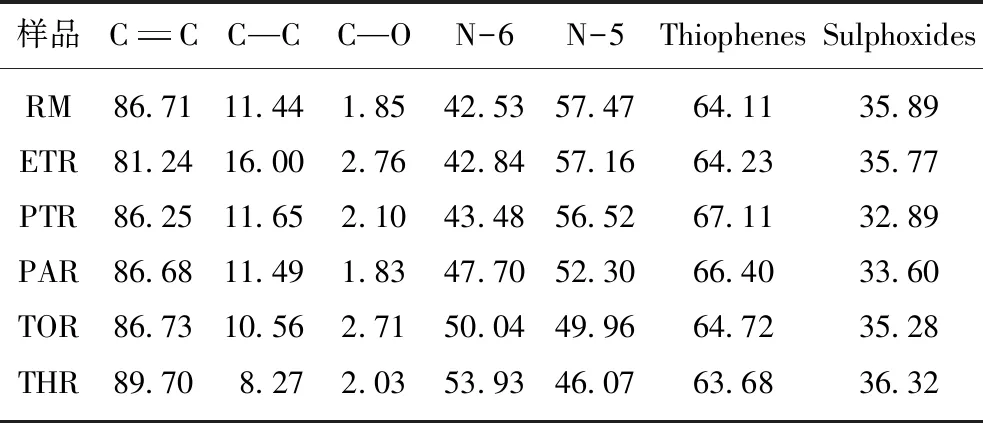
表4 原料及萃余物中C,N,S的不同形态及相对含量Table 4 Forms and the corresponding relative content of C,N and S in raw material and residues %
由图8可以看出,各级萃余物的C1s存在3种结合形态,结合能分别为284.6 eV 的C=C[37]、285.2 eV的C—C[38]和286.1 eV的C—O[39]。其中C=C的相对含量最高,超过80%,且与萃取级数正相关(表4),而C—C相对含量逐渐减少,说明随着萃取的进行,芳环的缩合度增加,特别是THR,C=C比例最高,C—C则最低。一般而言,N有5种形态,结合能分别为398.58,399.49,400.53,401.14和402.15 eV 处的吡啶型氮(N-6)、氨基氮、吡咯型氮(N-5)、质子化吡啶和氮氧化物[40]。图8显示萃余物中的N1s仅有N-6和N-5两个峰,意味着沥青中N以吡啶型和吡咯型存在。随萃取级数增加,吡咯型氮含量逐渐降低,一方面在于该类氮化合物更易萃取,另一方面则是由于萃取过程中各级萃余物结构单元内部的N-Q暴露在边缘,不能稳定存在亦可转化为吡啶氮[41],因而N-5的相对含量逐渐减小。沥青及萃余物的S1s含量极低(图8),可分为噻吩型和亚砜型[42](图8),其中前者比例较高,亚砜可能是源于煤沥青中芳硫的氧化[43]。
(4)晶相分析。图9为原料及各级萃余物的XRD谱图。图中仅在25°有一个(002)峰,且峰型较为弥散。在19°附近的γ峰可归结于芳环中环烷烃的衍射峰。表5给出了根据布拉格方程式计算各物质的微晶结构参数[44]。可见,随着萃取的进行,各物质的饱和链间距和芳香片层间距逐渐减小,芳香层片间直径、堆砌高度及芳香片层数逐渐增大,说明芳环的缩合程度增加,碳原子定向有序化增强,晶格化程度增大。
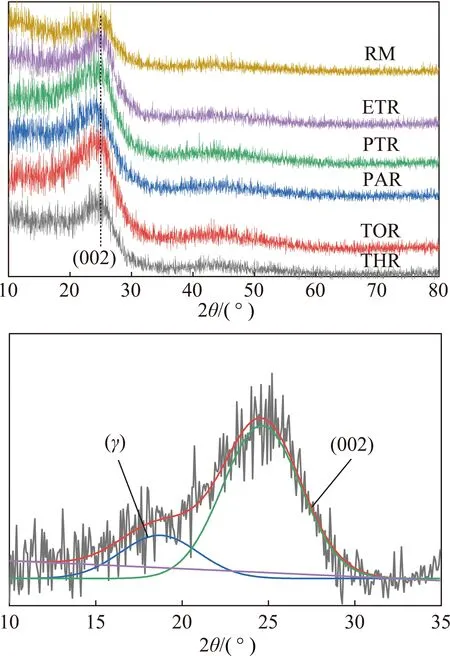
图9 原料及各级萃余物的XRD谱图及分峰拟合示意Fig.9 XRD spectrum and fitting curves of raw pitch and residues
(5)13C NMR分析。图10为中温煤沥青原料及各级萃余物的13C NMR谱图。可以看出,C主要以芳碳形式存在(90×10-6~170×10-6),在10×10-6~50×10-6存在微量的脂碳。由于各种碳峰之间有交叉重叠,通过Peakfit软件将谱图分峰拟合成6种不同类型的碳峰(图10(b)),相应的碳类型和结构参数及其相对含量[45]见表6。
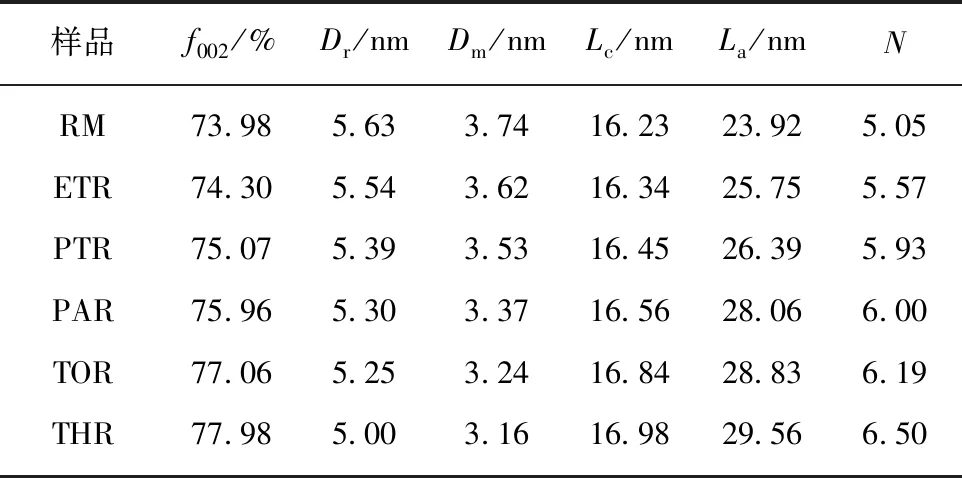
表5 原料及各级萃余物的微晶结构参数Table 5 Micro crystalline structure parameters of raw material and residues
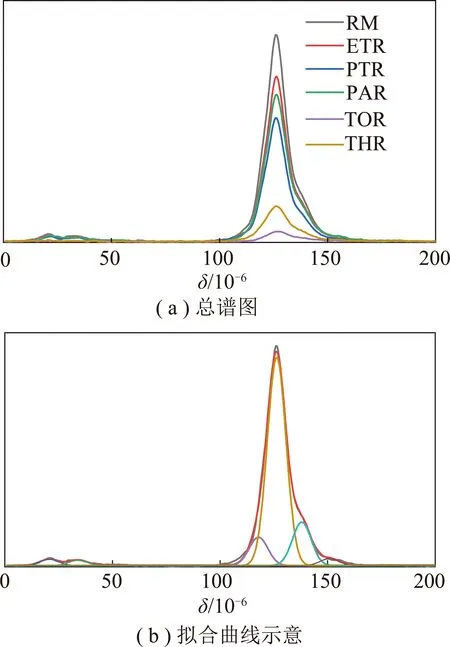
图10 样品的13C NMR分析Fig.10 13C NMR analysis of samples
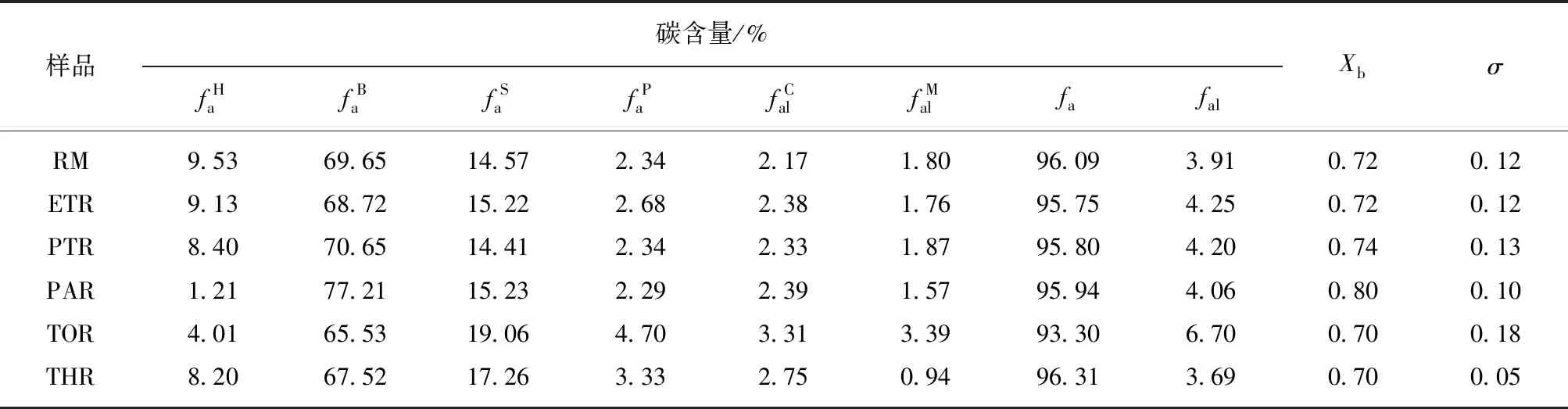
表6 原料及各级萃余物的固体13C NMR 的碳类型及碳结构参数Table 6 Different types of carbon and carbon structural parameters of solid-state13C NMR in raw materials and residues
3 结 论
(1)各萃取物分子量分布集中在100~400 u之间,以芳香烃和杂环化合物为主。
(2)C多以芳香C=C形式存在,N以吡啶-N和吡咯-N形式存在,S则以噻吩硫为主。
(3)随着萃取级数的增加,各级萃余物的热稳定性逐渐增强,各物质的饱和链间距和芳香片层间距逐渐减小,芳香层片间的直径、堆砌高度及芳香片层数逐渐增大。
(4)沥青及各级萃余物中平均芳环尺寸接近于5。
