S,D,L型解剖矫正型大动脉异位的产前超声心动图特征
2020-12-16李文秀方海燕
李文秀 方海燕 耿 斌 杨 爽 吴 江
解剖矫正型大动脉异位(ACMGA)是一种非常罕见的先天性心血管畸形,是由于圆椎动脉干发育异常,导致大动脉位置关系改变,但心室与大动脉的连接正常,即主动脉和肺动脉均起自相对应的心室[1]。ACMGA最常见的类型S,D,L,即心房正位,心室右襻,房室连接一致,大动脉连接正常,主动脉位于肺动脉左侧(即左侧异位);常合并室间隔缺损(VSD)、双动脉下或主动脉下圆锥、双侧心耳并置、左室流出道狭窄、主动脉弓异常等[2-7]。S,D,L型ACMGA十分罕见,胎儿期报道极少[8-10],极易被误诊为其他畸形。本文通过分析首都医科大学附属北京安贞医院(我院)儿童心血管病中心诊治及院外远程网络会诊的S,D,L型ACMGA胎儿的超声心动图图像,总结该畸形的超声特征,以期提高认识及诊断准确率。
1 方法
1.1 伦理和知情同意 本文通过我院伦理委员会审批(批准号:2020068X),所有参与研究的孕妇在胎儿超声心动图检查前均签署知情同意书。
1.2 纳入标准 ①2016年11月至2020年8月在我院儿童心血管病中心产前超声心动图检查或院外远程网络会诊诊断的S,D,L型ACMGA连续病例;②经胎儿尸体解剖或出生后经超声心动图、心脏CT或手术确诊。
1.3 临床资料截取
1.3.1 孕妇资料 ①年龄、产前超声检查时的孕周(由本次妊娠末次月经或妊娠早期超声检查确定)、受孕方式、单胎或多胎、孕期服药史及家族史;②外院和我院胎儿超声心动图诊断。
1.3.2 胎儿资料 ①心脏位置,心室比例;②心房是否正位、心房与心室连接是否一致;③2条大动脉与心室的关系;④2条大动脉是否存在包绕关系;⑤2条大动脉的空间位置;⑥2条大动脉下是否存在圆锥肌;⑦主动脉瓣与二尖瓣之间纤维连续是否存在,肺动脉瓣与三尖瓣之间是否为纤维连接;⑧是否合并其他的心脏畸形;⑨是否终止妊娠;⑩是否行手术治疗及手术后治疗效果。
1.4 胎儿产前超声心动图检查 使用Philips iE33、Epic 7C、GE Voluson E8彩色超声诊断仪,C5-1胎儿探头,频率2~5 MHz。选择胎儿心脏模式,按照Van Praagh节段分析法明确每个心脏节段的解剖状况及连接方式,最终确定心房位置、心房与心室连接方式、心室与2条大动脉连接关系及2条大动脉空间位置。重点扫查主动脉及肺动脉下有无肌性圆锥结构,二尖瓣与主动脉瓣、三尖瓣与肺动脉瓣是否为纤维连接,以及左、右心室流出道是否存在狭窄。扫查时常规使用CDFI或能量多普勒进行观察,并重点观察双侧心耳的位置及是否存在其他的心内畸形。
1.5 随访和验证 由我院儿童心血管病中心及院外的2~3名高年资医师共同做出产前诊断,向孕妇及其家属全面告知疾病的预后及转归,孕妇及家属自行选择继续或终止妊娠,终止妊娠胎儿建议引产后尸体解剖行病理检查,继续妊娠者于产后行新生儿超声心动图检查。
2 结果
2.1 一般情况 符合本文纳入标准的病例5例,孕妇年龄26~35(29.4±3.6)岁,孕周23~28(24.2±2.2)周;孕妇均为自然受孕、单胎,孕期无明确服药史及家族遗传史。表1显示5例胎儿及其母亲的一般情况,例3和5为我院产检病例,余3例为外院网络会诊病例,分别来自山东省潍坊市妇幼保健院、中国人民解放军东部战区总医院和福建省福州市第一医院。
表1显示,5例胎儿均合并其他的心内畸形,其中4例胎儿引产,3例家属拒绝尸检,例4尸检后证实产前超声所见;例5出生后2个月行心脏CT检查并行外科手术。

表1 5例S,D,L型ACMGA胎儿的临床资料、
2.2 病例报告
2.2.1 一般情况 例5,母亲35岁,自然受孕、单胎,在孕28周行超声心动图诊断,合并双侧心耳左侧并置,VSD,永存左上腔静脉(PLSVC),左室流出道轻度狭窄。胎儿出生后2个月行心脏CT检查并行外科手术,因年龄较小,1期先行肺动脉环缩术。
2.2.2 超声心动图和CT图像特征 孕28周和出生后2个月超声心动图(图1)、出生后心脏CT图像(图2)显示,①心房正位,心室右襻,房室连接一致(图1A、F);②大动脉平行发自相对应的心室,包绕关系消失(图1B、C、G);③主动脉位于肺动脉左前方,即左侧异位(图1B、G);④2条大动脉间可见圆锥肌(图1B);⑤主动脉瓣与二尖瓣之间为肌性连接(图1D);⑥肺动脉瓣与三尖瓣之间无圆锥肌(图1E);⑦左室流出道较长,形态似“天鹅颈”(图1D);⑧合并VSD、双侧心耳并置、PLSVC等其他心内畸形(图1E)。
3 讨论
ACMGA是由于圆锥动脉干发育异常,导致大动脉位置关系改变,但其与心室的连接正常,主动脉和肺动脉均起自相对应的心室。当心房正位时,右心房与右心室相连,位于右侧;左心房与左心室相连,位于左侧;2个心室具有正常的窦部结构,但2个心室流出道或漏斗部结构存在异常。主动脉瓣下一般都有圆锥肌,圆锥间隔发育良好,主动脉瓣环与二尖瓣环之间是肌肉组织,无纤维连续,右心室一般也有漏斗部,但常常发育不良。主动脉位于左侧并向前上移位,一般位于肺动脉的左前方。根据房室连接关系,Van Praagh等[1]将ACMGA分为4型,分别是:①S,D,L;②S,L,D;③I,L,D;④I,D,L。其中S,D,L型最为常见。部分学者在Van Praagh分型的基础上,将房室连接一致定义为ACMGA,而房室连接不一致的归于孤立性心室反位(IVI)[11-13]。

图1 例5孕28周和出生后2个月超声心动图图像
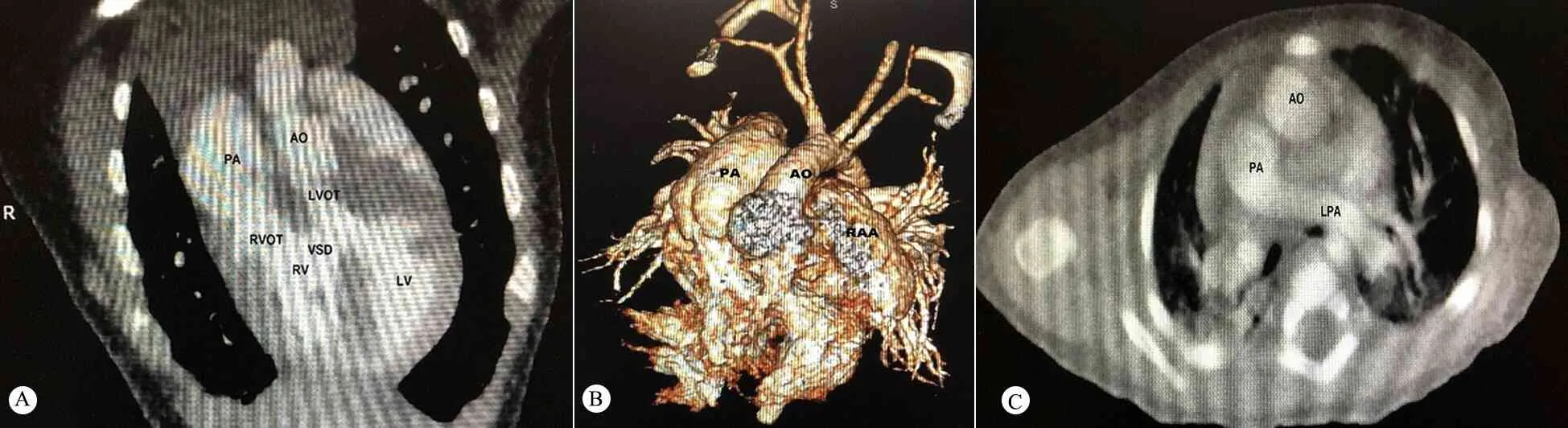
图2 例5出生后心脏CT图像
IVI也被称为房室连接不一致伴心室大动脉连接一致,或孤立性房室连接不一致,是心房与心室连接不一致而心室与大动脉连接一致的心血管畸形,较罕见,其特征为血流动力学改变和临床表现与完全型大动脉转位相同[14-16]。根据合并心内畸形的不同,选择的手术方式也不同,心房内调转术(Senning或Mustard术)是本病的根治手术,应用于早期不伴有明显肺动脉高压及严重肺动脉狭窄的病例[15]。
目前已有文献报道的ACMGA都合并其他先天性心脏畸形,包括膜周部VSD、双动脉下或主动脉下圆锥、双侧心耳并置、左室流出道狭窄、肺动脉狭窄、主动脉弓异常等[2-7]。由于房室连接和心室大动脉连接一致,ACMGA血流动力学正常,其手术方式主要取决于合并的畸形,多数患者需多期矫治手术[17-19];文献报道,S,D,L型的ACMGA术后生存率为92%,然而当房室连接不一致或合并右心发育不良时,ACMGA姑息手术的存活率仅为29%[11]。
本文例5出生后超声心动图及心脏CT检查结果与胎儿期检查结果一致。由于VSD对位不良,加之检查者对ACMGA畸形不了解,出生后在外院均被误诊为右室双出口(DORV),并根据VSD与大动脉关系,归类为主动脉下VSD型。DORV是本病需主要鉴别诊断的疾病,主动脉下VSD型DORV主动脉与肺动脉下存在圆锥肌,主动脉瓣环与二尖瓣环之间是肌肉组织,无纤维连续,但主动脉与肺动脉包绕关系存在,空间关系正常,即肺动脉位于左前、主动脉位于右后,与ACMGA的2条大动脉的空间关系完全不同。2种畸形的主要鉴别点为2条大动脉与房室瓣之间有无纤维联系及2条大动脉间的空间关系。该患儿由于年龄小,并存在肺动脉高压,为了避免肺动脉高压的进一步进展,1期行肺动脉环缩术,待患儿生长发育后再择期行VSD修补术及左室流出道狭窄矫治术。
文献报道,ACMGA可合并冠状动脉的异常,其中单支冠状动脉畸形最为常见[3,4,6];而冠状动脉的异常与主肺动脉的旋转有关,并影响手术方式的选择[20]。本文中,例5患儿出生后心脏CT及超声心动图检查均未发现冠状动脉异常,例4尸检后也未发现冠状动脉的异常。尽管胎儿期无法对此类冠状动脉的畸形进行明确诊断,但这可提示检查者应注意对出生后ACMGA患儿行冠状动脉扫查。
ACMGA除了应与DORV、IVI相鉴别外,还应与完全型大动脉转位、矫正型大动脉转位鉴别[8,9,11],在产前诊断的过程中,要多切面、多角度地扫查,并严格遵循Van Praagh的节段分析原则,明确每个心脏节段的解剖状况,确定各心脏节段之间的序列、房室连接方式及2条大动脉的空间关系,并最终做出准确的产前诊断,为产前咨询及产后的临床治疗提供依据。
