R/S型甲基苯丙胺药理活性差异的分子对接比较
2020-12-14崔露丹
崔露丹
摘 要: 甲基苯丙胺(MA)有一个手性碳原子,形成了R和S型分子。它们的物理性质(除偏振光外)、化学性质(除手性环境外)和分子光谱性质相同,但是它们的药理活性差异性大。用分子对接软件(Autodock 4.2.6)探讨手性R/S型甲基苯丙胺(MA)分子与单氧化聚合酶(DAT)结合能和结合位。结果表明:S-MA与DAT对接平均结合能量值为-34.03 kJ·mol-1,最低结合能量是-33.41 kJ·mol-1;而R-MA与DAT酶对接平均结合能量为-6.86 kJ·mol-1,最低结合能量是-5.60 kJ·mol-1,S-MA较R-MA对接DAT酶后形成的氢键数量多且强度大,而且结合能更负。与DAT结合效果最好的是S-MA,与DAT酶结合不稳定是R-MA。
关 键 词:R-甲基苯丙胺(R-MA)、S-甲基苯丙胺(S-MA);分子对接;单氧化聚合酶
中图分类号:TQ013 文献标识码: A 文章编号: 1671-0460(2020)10-2213-04
Abstract: Methamphetamine(MA) has a chiral carbon atom, forming R-type and S-type molecules. Their physical properties (except for polarized light), chemical properties (except for chiral environment) and molecular spectral properties are the same, but their pharmacological activities are quite different. Using Autodock 4.2.6 software and molecular docking software, the binding energy and binding sites of chiral R/S-methamphetamine (MA) with DAT enzyme were studied. The results showed that the average binding energy of S-MA to DAT was -34.03 kJ·mol-1, and the lowest binding energy was -33.41 kJ·mol-1, while the average binding energy of R-MA to DAT enzyme was -6.86 kJ·mol-1, and the lowest binding energy was -5.60 kJ·mol-1. Hydrogen bonds formed with S-MA were more and stronger than those formed with R-MA. The S-MA had the best binding effect with DAT, and the R-MA had unstable binding with DAT enzyme.
Key words: R-methamphetamine(R-MA); S-methamphetamine (S-MA); Molecular docking; DAT
甲基苯丙胺(MA)是一種新型的成瘾毒品,目前人们对其成瘾机制认知比较缺乏,治疗手段非常有限,涉及多种神经递质、受体和转运体,危害程度很高。20世纪70年代,美国、日本、德国等开始从行为学、形态学、分子水平对MA的作用机制进行了大量的研究,但一些机制并没有完全弄清楚,目前国内外的研究主要集中在氧化应激、神经元凋亡、胶质细胞活化等方面。王存新等对药物分子对接方法和蛋白质数据库进行细致的介绍,列举可以用于反向虚拟筛选的网络服务器,并列举该方法在药物设计领域的一些具体应用,最后对该方法目前所存在的问题进行讨论[1];谢治深等阐述了常用计算机合理药物设计方法及其在新药研发中的应用,为加快新药研发速度提供理论指导[2]。汪祺等考察芹菜素对UGT1A1酶活性的影响,从而预测其在临床用药时潜在的药物间相互作用,提示黄酮类母核结构具有与酶蛋白UGT1A1结合的特殊结构[3];袁凤珠等基于分子对接的HIV-1 RT抑制剂虚拟筛选的方法,探讨了核糖核酸酶活性[4];刘志强等就现阶段常用的网络药理学分析服务器、中药化学成分数据库、靶标预测技术等数据平台的使用方法、细节要点进行对比分析,并对其在中药网络药理学研究中的应用现状进行综述,以供研究人员参考[5];杨欣等基于网络药理学及分子对接分析熊果酸抗类风湿性关节炎的分子机制[6];白启峰介绍了Schrodinger药物虚拟筛选的基本原理、流程和使用方法,提高了药物发现的效率[7];肖泽云等从天然产物中筛选人类免疫缺陷病毒(HIV)非核苷类逆转录酶抑制剂(NNRTI)和整合酶链转移抑制剂作用位点的双靶点抑制剂[8];刘景陶和柳耀花通过分子对接筛选出大量具有生理活性的药物小分子,这些小分子作为先导化合物再经过多轮反复与受体靶标分子进行对接,不断优化结构,最终产生可能成药的候选物[9-10];刘燕茹针对当前甲状腺亢进症药物治疗存在的治愈时间长、且毒副作用大等问题,提出并实现了一种基于现有过氧化酶抑制剂甲亢平、甲巯咪唑、丙硫氧嘧啶以及甲硫氧嘧啶的新型TPO抑制剂的计算机辅助设计和虚拟筛选方法[11];黄溪等基于分子对接的选择性激素类药物的毒副作用研究,采用Sybyl-X软件中Surflex-Dock分子对接模块,评价选择性激素类药物与雌激素受体、孕激素受体、甲状腺激素受体、糖皮质激素受体、雄性激素受体的结合模式,以打分函数为标准,评价激素类药物与受体之间的亲和强弱关系,探讨药物与靶标的作用机制[12];史海龙等基于药物靶点从传统中药库中高通量虚拟筛选抑制剂,运用计算机虚拟筛选技术从传统中药数据库中寻找表皮生长因子受体激酶的中药小分子抑制剂[13];杜冉峰通过分子对接技术,以与衰老相关的雷帕霉素靶蛋白为对接受体,以茯苓、桑枝、木瓜、薏苡仁和厚朴5味祛湿药中的化学成分为对接小分子,初步筛选祛湿药中的抗衰老活性成分[14]。手性分子的物理性质完全相同(除了对偏振光的作用不同外),化学性质也完全相同(除了对手性试剂的作用不同外),但是它们的生物药理和毒理性差别很大,这个可能跟生物酶等大分子的手性有关,人们理解为手性差别导致空间位阻不同,导致化学性质不同。手性分子的这种性质差异,是否可以用第一性原理来计算出来呢?本文采用分子对接的方式来模拟小分子甲基苯丙胺手性配体与受体生物大分子ADT酶之间的相互作用,通过计算得知两者之间的最佳结合构象和最低结合能量,进行比较。宁中军等阐述了手性药物的概念、药理作用类型及研究制备方法[15]。奇云在《世界科学》杂志上介绍了2001年度诺贝尔化学奖的获奖成果对人们如何高选择性地引入和构建新的手性中心、以及如何控制产物的立体化学方面包括手性药物的研制与开发所起到的推动作用[16]。田四琦等说明了手性药物对映体药效学的立体选择性[17]。芮建中等阐述了手性药物对映体选择性的药物动力学和药效学与临床合理用药[18];曾苏等陈述了手性药物的药理及其体内对映体的测定方法[19];汪兵陈述了手性药物对药效的影响及立体选择性合成[20]。进一步探究它们之间的相互作用可为临床特异靶向治疗MA成瘾提供理论依据,也可为研发高效戒毒药物提供新思路。
1 实验部分
1.1 软件与模型
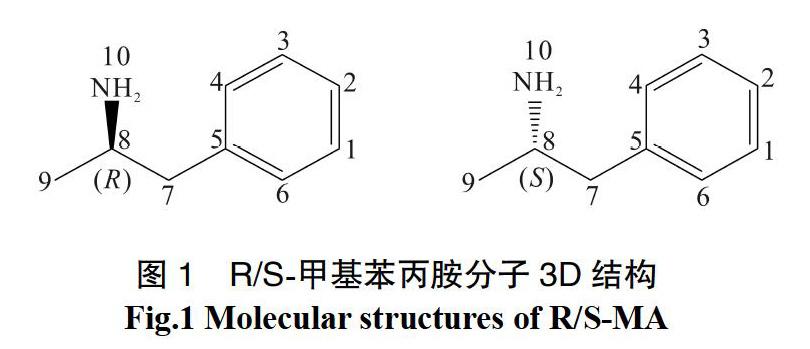
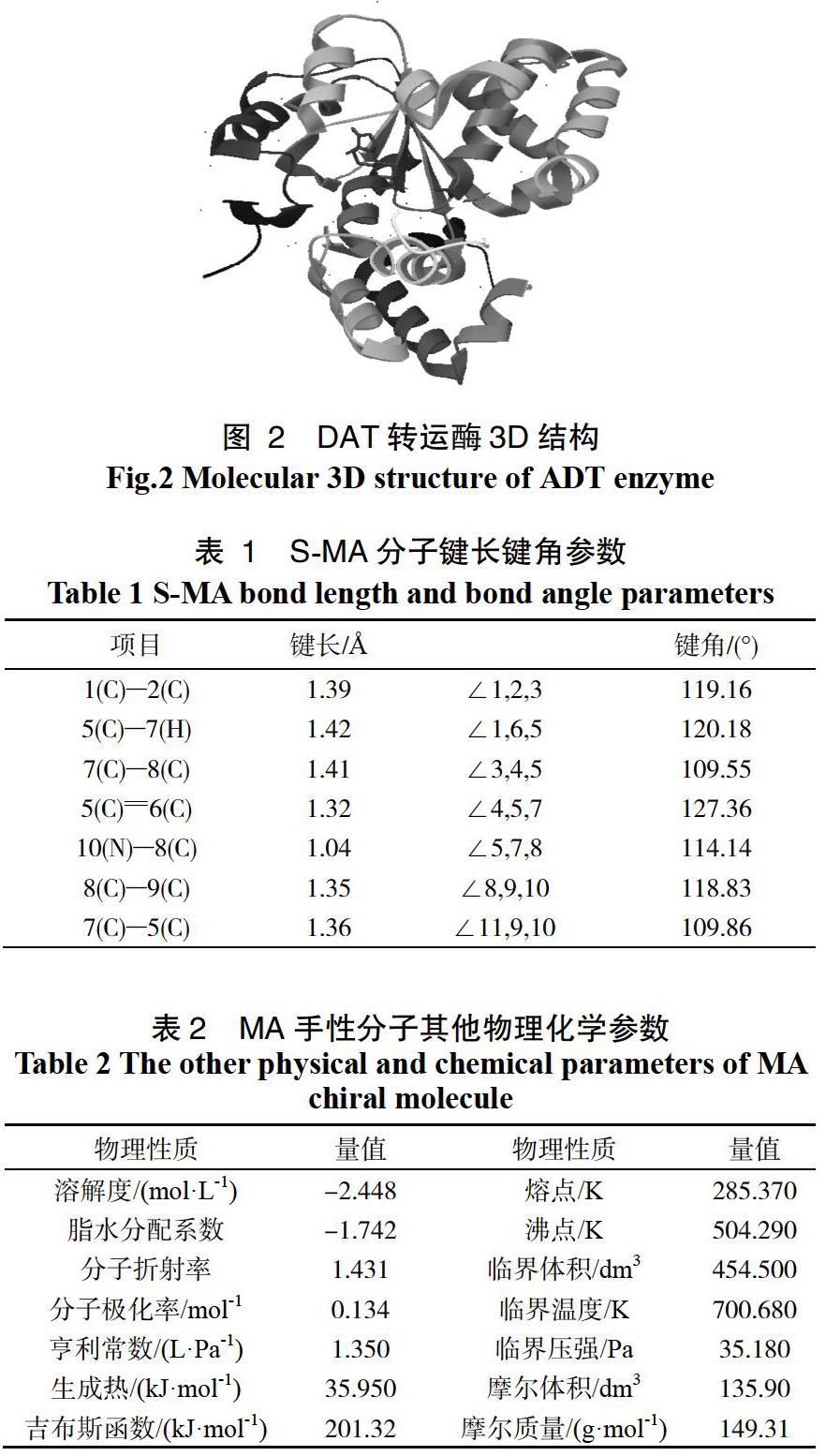
Chem3D、ChemDraw8.0、GaussView5.0.9、Gaussian09、AutoDockToos1.5.6等软件。Autodock是一款开源的免费分子对接模拟软件,AutoDock软件由 AutoGrid 和 AutoDock两个程序组成。其中 AutoGrid 主要负责格点中相关能量的计算,而 AutoDock 则负责构象搜索及评价。S型手性甲基苯丙胺的结构如图1所示。使用Chem3D软件,构建1个手性C原子模型,并使用ChemDraw8.0进行其能量最低化;在 GaussView 5.0.9 软件中,对其进行构型优化,并保存其 mol格式和gif 格式。突触前膜多巴胺转运体(DAT)酶3D结构式见图2。
1.2 分子骨架结构和性质对比
手性药物小分子MA的结构采用 Chemdraw 8.0进行建构与优化得到2D结构,再导入ChemBio3D软件得到3D结构,然后使用GaussView 5.0.9软件在B3LYP/-31G水平上进行结构优化。随后导入 AutoDockTools1.5.6 软件进行对接准备;突触前膜经多巴胺转运体(DA transporter,DAT)DAT酶的x线晶体结构是从蛋白质数据库(PDB)中提取的,删除PDB文件中所有的结晶水和配体分子。在对接之前我们需要将极性氢原子加上,与此同时科尔曼全原子电荷也要添加到酶的部分原子上,在进行对接时,需对酶进行刚性处理,而药物分子的键要设置为可自由旋转,以便进行半刚性对接。我们对每个体系都进行100次的对接,采用拉玛克遗传算法(LGA),最大能量评估为25 000 000。对于对接的结果处理,我们是根据均方根偏离标准 RMS= 2.0 ? 进行归类成簇,因为软件可以给出小分子与蛋白的结合自由能值;运用GaussView 5.0.9软件,分别选中两个原子进行键长计算,选中3个原子进行键角计算,S型甲基苯丙胺的键长键角计算结果见表1。由表1中数据可知,MA分子最长键长为碳碳单键1.42,最短键长为氧氢单键0.96 ?。最大键角为120.18,最小键角为109.86°。R-MA药物分子的骨架结构与其他性质完全相同(见表2)。多巴胺转运蛋白(DAT)是一种完整的膜蛋白,可從突触间隙中去除多巴胺并将其沉积到周围细胞中,从而终止神经递质的信号。它是由604个氨基酸残基构成,它含17个氨基残基的信号肽。它的C端有一个特异的18个氨基酸片段,能够使得其与膜结合(图2)。
2 结果与讨论
2.1 S-MA与DAT酶的对接结果
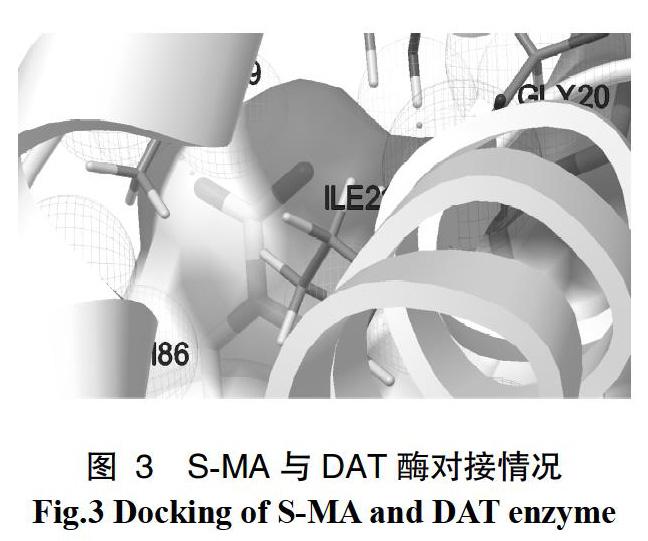
药物与蛋白质结合的稳定性跟其结合自由能大小有着紧密的联系,其结合自由能越负,小分子与蛋白的结合越牢靠;结合自由能越正,则小分子与蛋白结合越不牢靠,可以用来评价对接结果,使结果更具有可靠性。用AutoDock4.2.6软件进行分子对接。首先第一步打开AutoDockToos 1.5.6软件,在File菜单栏下找到Preferences Set打开文件夹用Make Default建立属于AutoDock的文档。第二步在Analyze菜单栏下找到Dockings Open打开小分子R/S MA的pdb格式文件,用Macromolecule Open打开大分子DAT的文件,用Clusterings Show进行分子结合。第三步是用Dockings Show Interactions进行分子对接。第四步选择最高能量柱,在Hydrogen Bonds菜单栏下找到Display As Lines得出分子对接氢键消息,根据氢键判断结合稳定性。根据对接软件得到的 S-MA与DAT酶对接图像如图3所示,S-MA分子与DAT对接后总共形成3个氢键。其中一个是S-MA分子上的氢与ILE20羰基上的氧形成的氢键;一个氢键是S-MA分子的氧与GLY20羰基上的氢所形成的, 另一个氢键是S-MA分子氧与IE80羰基上的氢所形成的。
通过Autodock软件计算可以得出它的具体对接数据见表3。由表3可以看出,数量最多的簇级是9号簇级,数量为12,平均结合能量为-8.07 kJ·mol-1,最低结合能量是 -8.14 kJ·mol-1,说明在S-MA与DAT酶对接中该簇级对接效果最好。通过对S-MA与酶的氢键数以及具体的对接数据分析,我们可以发现S-MA与DAT对接过程中第2簇族与第9簇族对接效果也是比较好的,说明S-MA药物对DAT酶具有较好的抑制作用。
2.2 R-MA与DAT酶对接结果
由图4可见,R-MA分子与DAT酶对接后总共只形成1个氢键。这个氢键是R-MA分子上的氢与DAT上的ALA96羰基上的氧形成的。而通过分子对接软件模拟计算得出的具体对接能的数据见表4。
由表4 可以看出,数量最多的簇级是2号簇级,数量为1,平均结合能量为-5.60 kJ·mol-1,最低结合能量是-6.85 kJ·mol-1,说明R-MA与DAT酶对接中该簇级对接效果好。
通过以上2组R/S手性分子MA虚拟对接实验,可以初步得出S-MA与DAT对接平均结合能量值为:34.03 kJ·mol-1,最低结合能是-33.73 kJ·mol-1;而R-MA与DAT酶对接平均结合能为: -6.86 kJ·mol-1,最低结合能量是-5.60 kJ·mol-1。
3 结 论
计算机辅助药物设计是计算机技术、分子生物学以及分子药理学等多学科交叉发展的基础上的一种药物研发新技术。将该方法应用于药物设计,这在很大程度上加快了药物设计和开发的速度。本文以计算机辅助药物设计的方法研究了两种手性药物分子S-MA型和R-M的物理化学性质差异以及它们分别与DAT单胺氧化酶的结合模式:
1)用AutoDock4.2.6软件进行分子对接,根据氢键以及最低结合能判断结合稳定性,最后得出MA分子与单氧化聚合DAT酶结合能与结合构象。将手性药物R/S-MA分别与转用DAT酶蛋白分子进行分子对接,通过分子动力学软件Autodock4.2.6模拟探究两复合物在真实环境中的稳定性差异。
2)选择AutoDockTools1.5.6分子对接程序,考察两种MA分子药物与ADT的结合构象。分子虚拟对接研究表明,S-MA和R-MA与DAT酶对接的两种情况中,S-MA存在3个氢键,R-MA只存在一个氢键。前者与DAT对接时所需要的最低结合能量最低,结合最稳定;S-MA与DAT酶对接时所需要的最低结合能量大,结合不稳定。所得结果为理解手性药物分子药理活性差异提供依据。利用对接方法研究新的抑制剂分子的对接结合模式以及对其结合能力进行分析的作用机制,将为防治吸毒的效用新靶点提供参考。
参考文献:
[1]王存新,许先进.基于分子对接的反向虚拟筛选方法[J].北京工业大学學报,2019,45(11):1164-1172.
[2]谢治深,宋军营,张振强,等.计算机辅助药物设计方法及其在新药研发中的应用[J].河南大学学报(医学版),2019,38(2):148-152.
[3]汪祺,王亚丹,杨建波,等.基于分子对接及体外抑制实验预测芹菜素潜在药物相互作用[J].中国中药杂志,2019,44(18):4043-4047.
[4]袁凤珠,肖向茜,曾毅.基于分子对接的HIV-1 RT抑制剂虚拟筛选的方法[J].中国艾滋病性病,2019,25(3):311-315.
[5]刘志强,王博龙.中药网络药理学药效成分筛选与靶标预测的研究进展[J].中成药,2019,41(1):171-178.
[6]杨欣,李亚辉,钱海兵,等.基于网络药理学及分子对接分析熊果酸抗类风湿性关节炎的分子机制[J].中国实验方剂学杂志,2018,24(18):207-214.
[7]白启峰,张洋,靳玲玲,等.Schr?dinger药物虚拟筛选流程模块在大学生物和化学信息学教学中的应用[J].大学化学,2018,33(5):66-71.
[8]肖泽云,李凯,李爱秀.中药化学数据库中HIVRT(NNRTI)/IN双靶点抑制剂的虚拟筛选[J].天津中医药,2018,35(5):386-391.
[9]刘景陶,柳耀花.通过分子对接确定并优化先导化合物[J].科技创新与应用,2018,11(3):3-4.
[10]刘景陶,柳耀花.计算机分子模拟技术及人工智能在药物研发中的应用[J].科技创新与应用,2018,11(2):46-47.
[11]刘燕茹,于敬达,李丽娜,等.新型TPO抑制剂的计算机辅助设计与虚拟筛选[J].科技通报,2017,33(12):230-232.
[12]黄溪,郑杰.基于分子对接的选择性激素类药物的毒副作用研究[J].计算机与应用化学,2017,34(8):639-644.
[13]史海龙,赵云飞,惠媛,等.基于药物靶点从传统中药库中高通量虚拟筛选EGFR-TK抑制剂[J].时珍国医国药,2016,27(9):2300-2304.
[14]杜冉峰,张小华,叶小彤,等.基于分子对接的祛湿药抗衰老活性成分筛选及作用机制解析[J].中国中药杂志,2016,41(13):2522-2526.
[15]宁中军,韩延峰,潘秀芹.手性药物的药理作用类型及其制备技术研究进展[J].中国医学创新,2009,6(25):194-196.
[16]奇云.开创手性催化反应研究的新领域[J]. 世界科学,2002,11 (1): 12-13.
[17]田四琦,刘会臣. 手性药物对映体药效学的立体选择性[J]. 药学实践杂志,1999,22(6):346-348.
[18]芮建中,吴锦芳,庞晓东.手性药物对映体选择性的药物动力学和药效学与临床合理用药[J].金陵医院学报,1998,13(1):66-71.
[19]曾苏,沈向忠,刘志强. 手性药物的药理及其体内对映体的HPLC测定[J]. 浙江医科大学学报,1994,12(5):234-238.
[20]汪兵.手性药物对药效的影响及立体选择性合成[J].齐鲁药事, 1992,15(2):23-26.
