5-氮杂-2'-脱氧胞苷对人子宫内膜样癌JEC细胞中P57kip2蛋白表达及细胞形态影响的研究
2020-12-11王凤月李三华刘俊江张春英周正平
周 俊,王凤月,李三华,钟 栎,赵 朔,刘俊江,张春英,周正平
(遵义医科大学1.病理学教研室;2.电镜室;3.医学与生物学研究中心;贵州 遵义 563099)
子宫内膜癌是女性生殖系统最常见的恶性肿瘤之一,其主要组织学类型是子宫内膜样癌(Endometrial carcinoma,EC)。2018年国际癌症研究中心(International Agency for Research on Cancer,IARC) 统计,全球约有382 069例新发子宫内膜癌患者,有89 929例患者死亡,其高发病率和高死亡率严重威胁着女性健康。目前研究发现,无孕激素拮抗的高水平雌激素刺激、肥胖、高血压、糖尿病、不孕、绝经期晚等均为子宫内膜癌的高危因素。但导致子宫内膜癌发生发展的确切机制尚不清楚,可能还与基因突变、翻译后修饰等有关[1-3]。
p57kip2是最迟被发现的细胞周期调控抑制因子,能广泛抑制细胞周期蛋白依赖性激酶 (Cyclin-dependent kinase,CDK)的激酶活性,使细胞无法从G1期转变到S期,起到负性调控细胞周期的作用[4]。目前研究发现,在多种肿瘤组织中,P57kip2蛋白水平的异常表达可能与表观遗传修饰有关[5]。DNA甲基化是基因表观遗传修饰的重要方式,主要发生于基因启动子区CpG岛,在不改变DNA碱基序列的前提下,通过甲基转移酶(DNA methyltransferase,DNMT)的催化作用,使CpG岛5’端的胞嘧啶变成5’甲基胞嘧啶(5-mc),从而改变肿瘤细胞的表型,进而影响肿瘤相关基因的表达[6]。5-Aza-CdR是一种DNMT抑制剂,可使DNMT失活,从而使高甲基化状态的肿瘤抑制基因启动子区DNA去甲基化,激活肿瘤抑制基因,使其重新恢复表达,发挥其抑制肿瘤细胞增殖、促进细胞分化的功能[7]。本实验拟选用人EC的中分化JEC细胞,明确细胞中p57kip2基因变异情况;用5-Aza-CdR干扰后,检测JEC细胞中p57kip2基因启动子区甲基化水平及其蛋白表达情况、细胞生长及细胞形态变化,探讨p57kip2对子宫内膜癌增殖、分化的的影响。
1 材料与方法
1.1 细胞株 中分化人EC JEC细胞株由遵义医科大学微生物教研室提供。
1.2 试剂与仪器 5-Aza-CdR购自MACKLIN公司,MTT 相关试剂购自南京凯基公司,WB相关试剂均购自上海碧云天生物研究所,p57kip2兔抗人单克隆抗体、二抗山羊抗兔抗体购自Abcam公司,RPMI 1640培养基、南美胎牛血清购自美国Gibco公司。CO2细胞培养箱、酶标仪(美国Thermo Scientific公司),倒置显微镜DM4000B (日本OLYMPUS公司),透射电子显微镜H-7650(日本日立公司),扫描电子显微镜FE-SEM SU8010(日本日立公司),Odyssey双色红外激光成像系统(美国LI-COR公司)。Sanger测序、BSP检测均由生工生物工程(上海)股份有限公司完成。
1.3 方法
1.3.1 Sanger测序法检测p57kip2基因外显子是否存在突变 取对数生长期细胞,消化离心后送生工生物工程(上海)股份有限公司检测。引物设计:F,5′-CCGTGGTGTTGTTGAAACTGAA-3',R,5' GTCCACTGCAGGCGTCCA-3',636 bp。采用PCR仪分别对p57kip2基因进行扩增。PCR 50 μL反应体系:模板1 μL,引物Fi 1 μL,引物Ri 1 μL,dNTP10 mmol/L 1 μL,Taq Buffer 5 μL,25 mmol/L MgCl25 μL,5 U/μL Taq酶0.5 μL,水35.5 μL。PCR反应条件:95 ℃预变性3 min,94 ℃变性30 s,55~60 ℃退火35 s,72 ℃延伸40~50 s,72 ℃修复延伸5~8 min,上述条件循环35次后1%琼脂糖及150 V、100 mA 20 min电泳后使用Sanger测序法检测p57kip2基因外显子区域,即对突变位点进行PCR扩增后进行测序验证测序。
1.3.2 BSP法检测JEC细胞中p57kip2基因启动子区甲基化情况 结合查阅相关文献[8-9],5-Aza-CdR浓度以10 μmol/L为参考设置浓度梯度(0、5、7.5、10、12.5、15 μmol/L),用等量含药培养基干扰JEC细胞24 h,交由生工生物工程(上海)股份有限公司提取DNA进行BSP检测p57kip2基因启动子区32个CpG位点的甲基化情况。引物设计,F,5′-GTTAGTAGGTGTGTGAGGGTTTTAG-3′,R,5′-CTCCTTTATCTACAAACRAAAACCTC-3′,298 bp。PCR反应条件:95 ℃预变性3~5 min,94 ℃变性30 s,55~60 ℃变性25~30 s,72 ℃退火30~50 s,上述条件循环35次,最后72 ℃修复延伸5~8 min后测序。
1.3.3 细胞培养及实验分组 将RPMI 1640培养基、南美胎牛血清和100 U/mL双抗按90%、10%和1%的比列配制成完全培养基,37 ℃、5% CO2、饱和湿度条件下体外培养人EC JEC细胞。根据BSP检测结果,选择甲基化率下降明显的两个5-Aza-CdR浓度(12.5 μmol/L 和15 μmol/L)的培养基体外培养JEC细胞作为实验组,等量完全培养基培养作为对照组。
1.3.4 MTT法检测JEC细胞增殖 取对数生长期细胞制成1×105个/mL的单细胞悬液,以100 μL/孔接种于96孔板上,待细胞贴壁后,将实验组和对照组分别培养24、48、72 h后,PBS冲洗每孔,加20 μL、5 mg/mL MTT溶液,至孵箱反应4 h后吸出MTT溶液,加100 μL/孔二甲基亚砜( DMSO),避光震荡10 min,用酶标仪于波长490 nm处检测各组的吸光光度值(OD值) 。
1.3.5 光镜、电镜观察JEC细胞形态结构变化
1.3.5.1 倒置显微镜观察JEC细胞生长情况 取对数生长期细胞制备成1×106个/mL的单细胞悬液,按接种于6孔板上,待细胞贴壁,将实验组和对照组分别培养72 h后于倒置显微镜下观察各组细胞的生长情况。
1.3.5.2 电镜观察JEC细胞超微结构 将实验组和对照组JEC细胞分别培养72 h后,消化、离心,将细胞团块进行两次固定、梯度脱水。浸透、包埋、聚合,半薄切片定位、超薄切片、双重染色后于透射电镜(Transmittance Electron Microscopy,TEM)下观察细胞内部超微结构;临界点干燥,粘托,镀膜后于扫描电镜(Scanning Electron Microscopy,SEM)下观察细胞表面超微结构。
1.3.6 Western blot法检测JEC细胞中P57kip2蛋白表达 将实验组和对照组分别培养24 h、72 h,加裂解液置冰上裂解30 min,提取蛋白后用BCA法测蛋白浓度,根据试剂说明书配置合适的分离胶和浓缩胶,5×上样缓冲液:蛋白按1∶4的比例混匀后沸水煮3~5 min,进行上样、电泳,切下p57kip2蛋白分子量大小所在区域的凝胶转膜、封闭,一抗(稀释相应倍数的β-actin、p57kip2兔抗人单克隆抗体)4 ℃摇床孵育过夜,洗膜3次,二抗(稀释相应倍数的山羊抗鼠抗体)室温摇床孵育2 h,洗膜3次,于Odyssey系统扫描成像,检测目的蛋白的相对含量(P57kip2蛋白灰度值/β-actin灰度值)。

2 结果
2.1 JEC细胞中p57kip2基因突变情况 在JEC细胞中,p57kip2基因外显子组测序对比后结果显示无突变(见图1)。
图1 p57kip2基因外显子测序峰图
2.2 JEC细胞中p57kip2基因启动子区甲基化水平 在JEC细胞中,p57kip2基因启动子区呈高甲基化状态(见图2),经各浓度5-Aza-CdR(5、7.5、10、12.5、15 μmol/L)干扰24 h后,其总体甲基化率(分别为88.1%、83.8%、85.8%、76.2%、83.1%)均低于0 μmol/L(88.8%),其中12.5 μmol/L组(76.2%)、15 μmol/L组(83.1%)的p57kip2基因部分位点去甲基化效果显著,总体甲基化率下降明显。和对照组比较,12.5 μmol/L组总体甲基化水平下降最明显。
2.3 5-Aza-CdR干扰后,JEC细胞的生长情况 相同作用时间:和对照组比较,12.5 μmol/L组细胞生长水平在24、48 h被明显抑制(P<0.05),72 h抑制不明显(P>0.05);15 μmol/L组在48 h明显抑制(P<0.05),24 h和72 h抑制不明显(P>0.05);在24、48 h,12.5 μmol/L组细胞生长水平均明显低于15 μmol/L组(P<0.05),在72 h两实验组之间则无明显差异(P>0.05)(见图3)。
相同药物浓度:随着5-Aza-CdR作用时间的延长,对照组和实验组细胞生长水平呈上升趋势。和24 h比较,12.5 μmol/L组在48 h和72 h上升均较为平缓(P>0.05);15 μmol/L组在48 h上升较平缓(P>0.05),72 h则明显上升(P<0.05)(见图3)。
2.4 5-Aza-CdR干扰后,JEC细胞中P57kip2蛋白的表达情况 相同作用时间:和对照组比较,在24 h,P57kip2蛋白在两实验组的表达均有所上调,且12.5 μmol/L组上调水平高于15 μmol/L组(P<0.05),但仅在12.5 μmol/L组的表达上调有意义(P<0.05);在72 h两个实验组均无明显上调,两实验组之间比较亦无明显差异(P>0.05,见表1,图4)。
相同药物浓度:随着时间延长,P57kip2蛋白在对照组中的表达变化不明显(P>0.05)。在两实验组中的表达均呈下降趋势,但仅12.5 μmol/L组中的表达下调显著(P<0.05),15 μmol/L组无明显下调(P>0.05,见表1,图4)。

图2 5-Aza-CdR干扰24 h后JEC细胞p57kip2基因启动子区甲基化情况
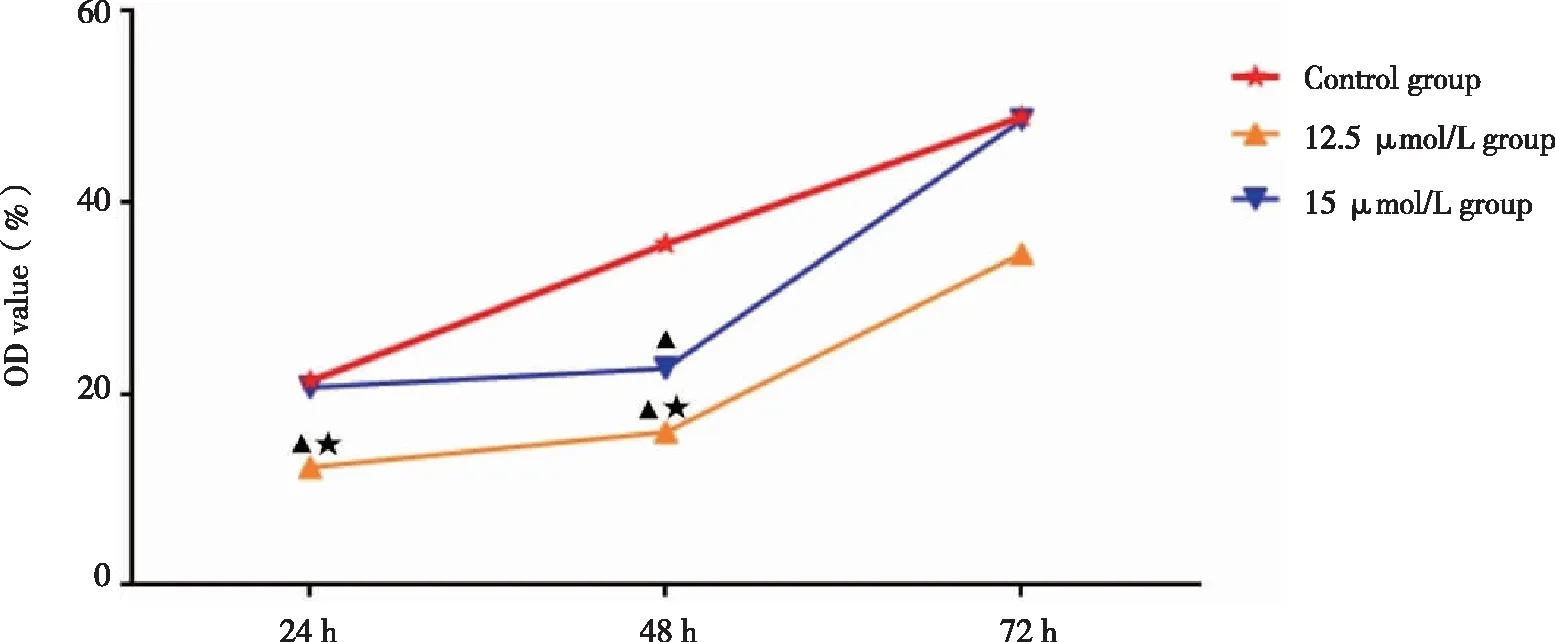
▲:与对照组比较,P<0.05;★:两实验组之间比较,P<0.05。图3 5-Aza-CdR干扰JEC细胞的OD值
表1 5-Aza-CdR干扰JEC细胞24、72 h后P57kip2蛋白表达
2.5 5-Aza-CdR干扰72 h后JEC细胞的形态变化
2.5.1 倒置显微镜观察JEC细胞的形态变化 对照组JEC细胞主要呈多边形或长梭形,可见明显的围腺腔样结构(见图5)。12.5 μmol/L组较对照组和15 μmol/L组细胞密度略微减少,15 μmol/L组细胞密度较对照组无明显差异,未见明显细胞形态改变。
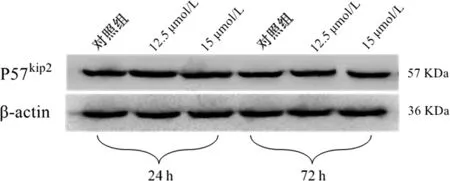
图4 5-Aza-CdR干扰24、72 h后P57kip2蛋白在JEC细胞中的表达情况

A:对照组;B:12.5 μmol/L组;C:15 μmol/L组。图5 3组JEC细胞72 h后的细胞生长情况
2.5.2 TEM和SEM观察JEC细胞的超微结构变化 对照组:JEC细胞多数呈椭圆形,细胞游离面可见微绒毛;细胞内部可见线粒体、内质网、自噬溶酶体散在分布于胞质内,还可见分泌泡、脂滴,细胞核大,核仁明显,核膜齿状曲折(见图6)。
和对照组比较,5-Aza-CdR干扰72 h后,两浓度组JEC细胞表面的微绒毛更加丰富,分泌泡明显增多;部分细胞内质网扩张明显,个别细胞内自噬现象更明显,未见明显线粒体肿胀(见图7~8);两浓度组细胞形态改变无明显区别。
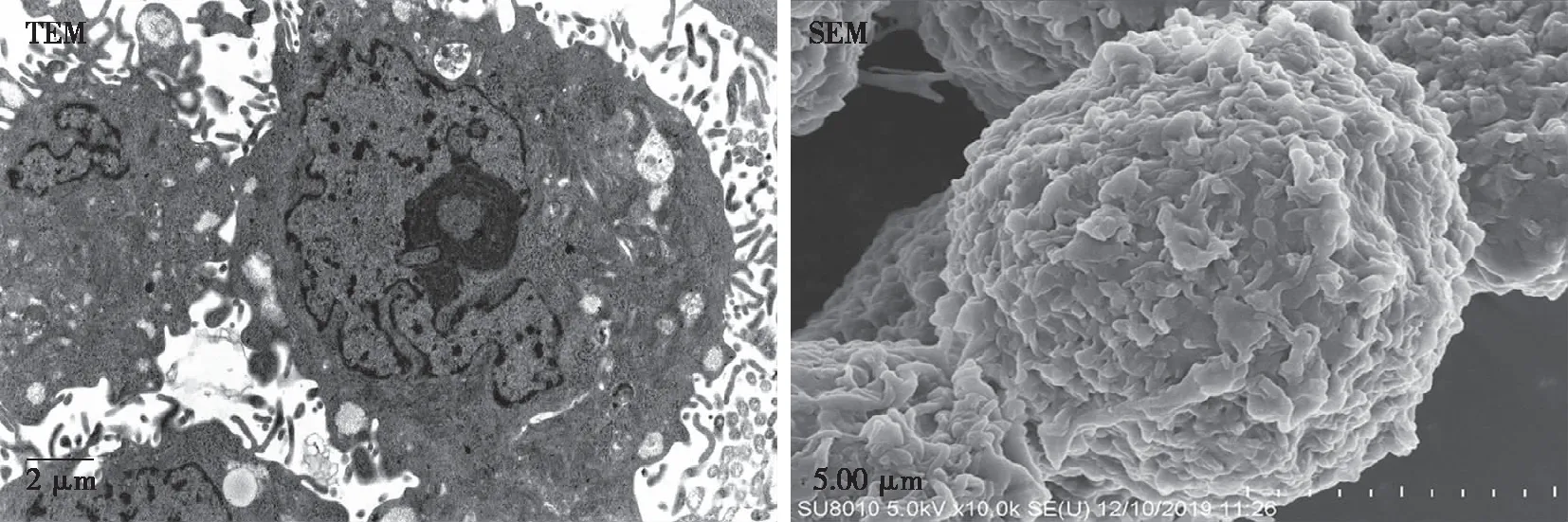
图6 对照组JEC细胞72 h后的细胞超微结构
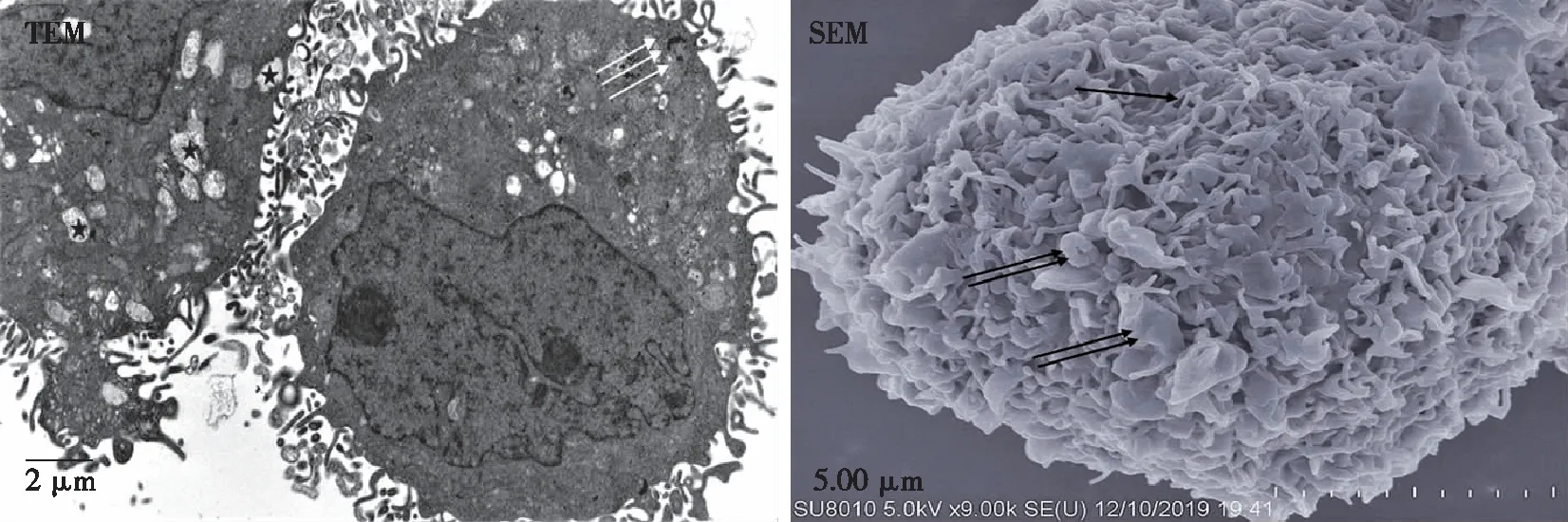
微绒毛(↑),分泌泡(↑↑,★),自噬现象(↑↑↑)。图7 12.5 μmol/L组JEC细胞72 h后的细胞超微结构
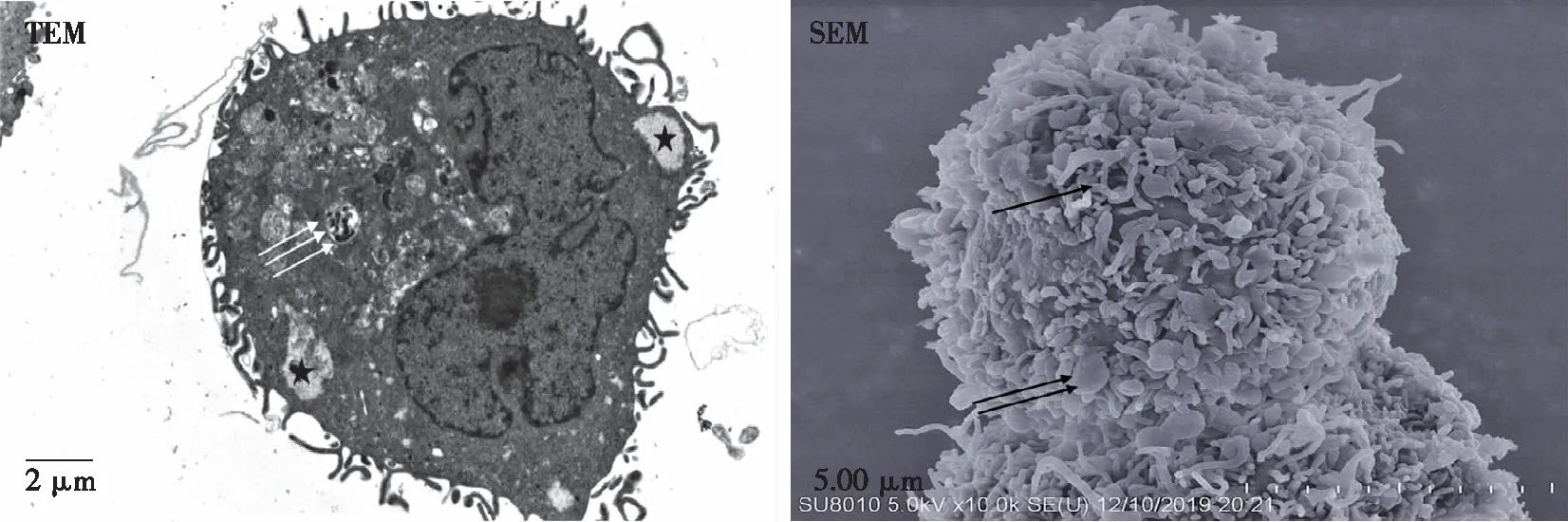
微绒毛(↑),分泌泡(↑↑,★),自噬现象(↑↑↑)。图8 15 μmol/L组JEC细胞72 h后的细胞超微结构
3 讨论
子宫内膜癌变是一个复杂的过程,肿瘤抑制基因失活、癌基因激活、DNA修复异常等参与其中。DNA启动子高甲基化是子宫内膜癌中肿瘤抑制基因失活的重要机制之一[10]。
p57kip2是最迟被发现的细胞周期依赖性激酶抑制剂,位于染色体11p15.5,包含4个外显子和3个内含子,全长22 kb,其编码的蛋白质分子量为572 kDa,由316个氨基酸构成[11]。p57kip2作为一种候选的肿瘤抑制基因,在乳腺癌、前列腺癌、肝癌等组织中表达明显下调或缺乏,其表达水平较高时预示着肿瘤的分化程度较高、临床分期较早、淋巴结转移率较低、预后较好[12-14]。本研究发现,在JEC细胞中,p57kip2基因外显子组未发生突变,提示p57kip2蛋白在子宫内膜样癌细胞中的异常表达可能与表观遗传修饰中DNA甲基化等有关。
表观遗传学修饰是在不改变DNA碱基序列的前提下,改变基因的表型,进而使基因功能发生可遗传的变化,主要有DNA甲基化(DNA methylation)、组蛋白修饰(Histone modification)、染色体重塑(Chromatin remodeling) 和小RNA(MicroRNA) 调控等表现形式[15]。DNA甲基化是一个动态的、可逆转的调节过程,可通过抑制DNMT酶活性、脱甲基、酶促5-mc降解等方式阻止甲基化过程,使DNA在复制循环的过程中逐渐去除DNA甲基化[16-17]。我们研究发现,在未行干预的JEC细胞中,p57kip2基因启动子区呈高甲基化状态。提示在子宫内膜癌组织或细胞中,p57kip2基因启动子区高甲基化状态可能导致p57kip2对细胞增殖的负调控作用被抑制。
5-Aza-CdR又名地西他滨,是目前体内外用于抑制DNA甲基化最常用的药物,能使DNMT失活来抑制甲基化过程,从而使处于高甲基化状态的基因去甲基化、恢复抑癌基因的功能[18]。吴晓玲等[19]发现,宫颈癌组织中SOX11基因(被认为是一种抑癌基因)沉默与其启动子区高甲基化密切相关,经去甲基化药物5-Aza-CdR处理后,SOX11恢复表达。黄丽萍等[8]研究显示,子宫内膜癌HEC-1-B细胞经5-Aza-CdR处理后,RASSF1A基因及其蛋白重新表达,发挥其抑制细胞增殖、侵袭的能力,且5-Aza-CdR对RASSF1A基因去甲基化及其蛋白表达增加的调控具有时间和浓度依赖性。Yang等[9]发现,使用5-Aza-CdR处理人结肠癌HCT116细胞后,能快速抑制细胞生长和延长细胞倍增时间,在没有进一步加药治疗的情况下,细胞生长抑制状态会逐渐逆转,使用HM450 DNA甲基化阵列长期监测DNA甲基化的变化,发现不同位点的再甲基化率不一致。
我们研究发现:JEC细胞经各浓度5-Aza-CdR(5、7.5、10、12.5、15 μmol/L)干扰24 h后,其总体甲基化率(分别为88.1%、83.8%、85.8%、76.2%、83.1%)均低于对照组(88.8%),其中12.5 μmol/L组(76.2%)、15 μmol/L组(83.1%)的p57kip2基因部分位点去甲基化效果显著,总体甲基化率下降明显。和对照组比较,12.5 μmol/L组总体甲基化水平下降最明显。和对照组比较,两实验组P57kip2蛋白表达均有上调,细胞生长均被抑制,但以12.5 μmol/L组最明显。随着5-Aza-CdR作用时间延长,两实验组P57kip2蛋白表达有所下降、细胞生长水平有所增长,这与Yang等[9]的研究相似、与黄丽萍等[8]的研究不一致,可能与没有持续追加5-Aza-CdR维持去甲基化水平有关,还可能与肿瘤的不同组织类型、不同细胞系对5-Aza-CdR的敏感性及其内在基因组表观特征不同有关;我们还发现,两实验组较对照组细胞密度略有减少、细胞表面微绒毛均更丰富,细胞内分泌泡和自噬现象均增多。由此可以得出,在JEC细胞中,P57kip2处于高甲基化状态,使其负向调控细胞进程的作用被减弱,细胞明显增殖。5-Aza-CdR干预后,可下调JEC细胞中p57kip2基因启动子区甲基化水平,上调P57kip2蛋白表达,激发其负向调控细胞周期的作用,从而抑制JEC细胞增殖,其中12.5 μmol/L组的生长抑制作用比15 μmol/L组更明显,可能还使JEC细胞往更好的分化方向转变;随着时间的延长,没有进一步追加5-Aza-CdR,p57kip2启动子区逐渐恢复甲基化,导致P57kip2蛋白表达上调的趋势再次被减弱,细胞生长的抑制状态也会被逆转。5-Aza-CdR的去甲基化效果可能需要持续加药来维持,将是下一步实验需要验证的问题。
