铁死亡诱导剂和抑制剂的研究进展
2020-12-09孙晟杰唐励静罗秀菊
孙晟杰,涂 画,唐励静,罗秀菊,彭 军
(1.中南大学湘雅药学院药理学系,湖南 长沙 410078;2.中南大学湘雅医学院检验系,湖南 长沙 410013)
2012年,DIXON 等[1]发现 erastin可以促进Ras突变的癌细胞死亡,触发一种独特的调节性坏死,表现为细胞内铁离子累积、脂质活性氧(reactive oxygen species,ROS)增多和线粒体膜密度增加等特点,因这种新型细胞死亡方式具有依赖铁离子的特点,故命名为铁死亡(ferroptosis)。
铁死亡的发生与铁、氨基酸、谷胱甘肽(glutathione,GSH)、ROS和脂质过氧化物(lipid peroxidation,LPO)等代谢有关。细胞外液的Fe3+通过转铁蛋白转运至细胞内并被还原为Fe2+,Fe2+与细胞内过量的H2O2通过芬顿反应(Fenton reaction)生成大量ROS,促进细胞内LPO的生成而触发铁死亡[2]。参与铁死亡的信号通路包括胱氨酸/谷氨酸反向转运体(SystemXc)途径、铁稳态调节途径、电压依赖性阴离子通道(voltage-dependent anion channel,VDAC)途径、磷酸戊糖途径[1-2]、甲羟戊酸途径[3]、腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)-BECN1[4]和 kelch 样 ECH 相关蛋白 1(kelch-like ECH-associated protein 1,KEAP1)-核因子E2相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)-血红素加氧酶1(heme oxygenase 1,HMOX1)[5]等通路,这些信号通路相互交集,发挥综合效应(图1)。
铁死亡的发生使细胞产生独特的形态学改变:细胞变成圆形并相互分离,线粒体体积缩小、内膜凝聚、嵴皱缩或消失和外膜破裂,但细胞核结构完整,无凝聚或染色质着边现象,这些形态学特征有助于区分铁死亡与其他形式的调节性坏死[1-2]。
近期研究表明,铁死亡对肿瘤、心肌梗死、卒中和神经退行性疾病(如帕金森病)等多种疾病的发生发展具有重要意义,通过人为干预诱导或抑制铁死亡,将为这些疾病的治疗提供新思路。目前已发现多种化合物可诱导或抑制铁死亡发生,称为铁死亡诱导剂或抑制剂。本文旨在从发现、结构、应用模型、作用靶点和作用特点等方面对铁死亡诱导剂和抑制剂分别进行总结归纳,重点论述经典铁死亡诱导剂和抑制剂等的研究进展,对新近发现的具有铁死亡诱导和抑制作用的其他化合物和天然产物也进行简要综述,为其进一步研究、开发和临床应用提供理论参考。
1 铁死亡诱导剂
铁死亡诱导剂的化学结构见图2,按作用靶点将铁死亡诱导剂分为4类(表1):靶向胱氨酸/谷氨酸反向转运体、靶向谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)、靶向GSH以及靶向铁离子和ROS的诱导剂。
1.1 靶向胱氨酸/谷氨酸反向转运体
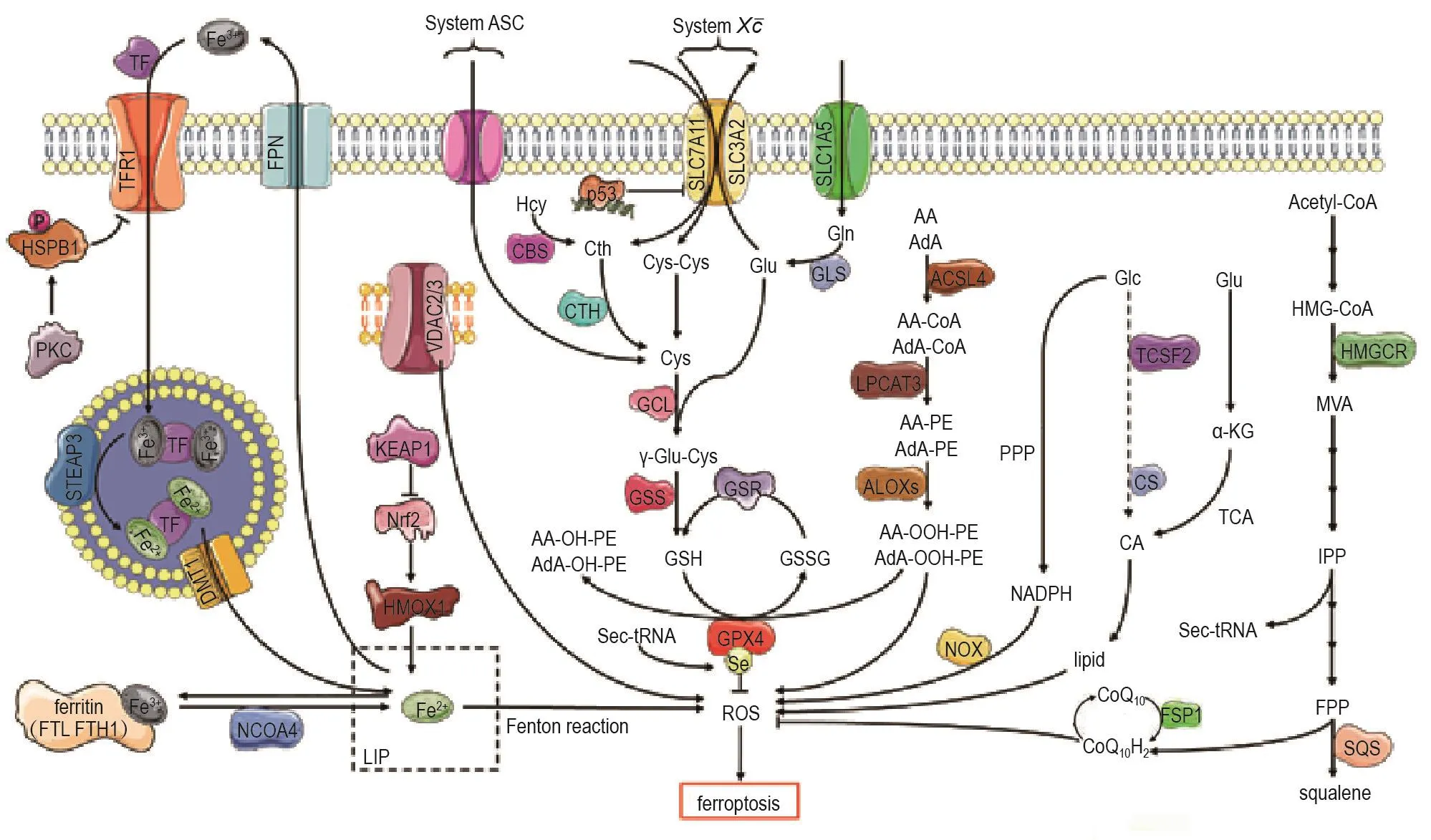
图1 铁死亡的主要信号通路和关键调节因子.TFR1:转铁蛋白受体1;PKC:蛋白激酶C;HSPB1:热休克27 ku蛋白1;STEAP3:前列腺6次跨膜上皮抗原3;DMT1:二价金属转运蛋白1;FTL:铁蛋白轻链;FTH1:铁蛋白重链1;NCOA4:核受体辅激活因子4;LIP:不稳定铁池;HMGCR:3-羟基-3-甲基戊二酰辅酶A还原酶;ACSF2:酰基辅酶A合成酶家族成员2;CS:柠檬酸合成酶;NOX:NADPH氧化酶;LPCAT3:溶血磷脂酰胆碱酰基转移酶3;SLC1A5:溶质载体家族1成员5;System ASC:丙氨酸/丝氨酸/半胱氨酸转运体;System X c-:胱氨酸谷氨酸反向转运体;CBS:胱硫醚-β-合成酶;CTH:胱硫醚酶;GLS:谷氨酰胺酶;GSS:谷胱甘肽合成酶;GSR:谷胱甘肽还原酶.

图2 部分铁死亡诱导剂的化学结构.
SystemXc由轻链溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)和重链溶质载体家族3成员2(solute carrier family 3 member 2,SLC3A2)组成,SLC7A11是反向转运体的主要活性亚基,调控细胞内GSH的动态平衡。抑制SystemXc-的活性可减少胱氨酸转运入胞,使胞内GSH合成减少,降低GPX4清除过氧化物的能力,细胞内累积的LPO增多,同时引发内质网应激反应和Chac1基因上调,最终导致细胞铁死亡[1,6]。
1.1.1 erastin及其衍生物
erastin是最早发现的铁死亡特异性诱导剂(图2-1),可直接抑制SystemXc的活性,影响GSH的合成,最终导致人纤维肉瘤细胞HT-1080、人包皮成纤维细胞BJeLR和人肺癌细胞Calu-1等发生铁死亡[1]。亲和纯化和质谱分析表明,VDAC2/3也是erastin的直接靶点之一,用shRNA沉默Vdac2/3可引起HT-1080细胞对erastin的显著抗性,erastin-VDAC2相互作用降低VDAC2对烟酰胺腺嘌呤二核苷酸的通透性,并可能改变其离子选择性,允许阳离子进入,使线粒体去极化,诱导线粒体功能障碍,引起ROS的生成增多,也能促进细胞铁死亡[7]。分子构效关系显示,erastin的活性中心是喹唑啉酮部分,哌嗪连接基的刚性减弱会降低其活性,氯原子是其与周围环境相互作用的重要位点[6]。
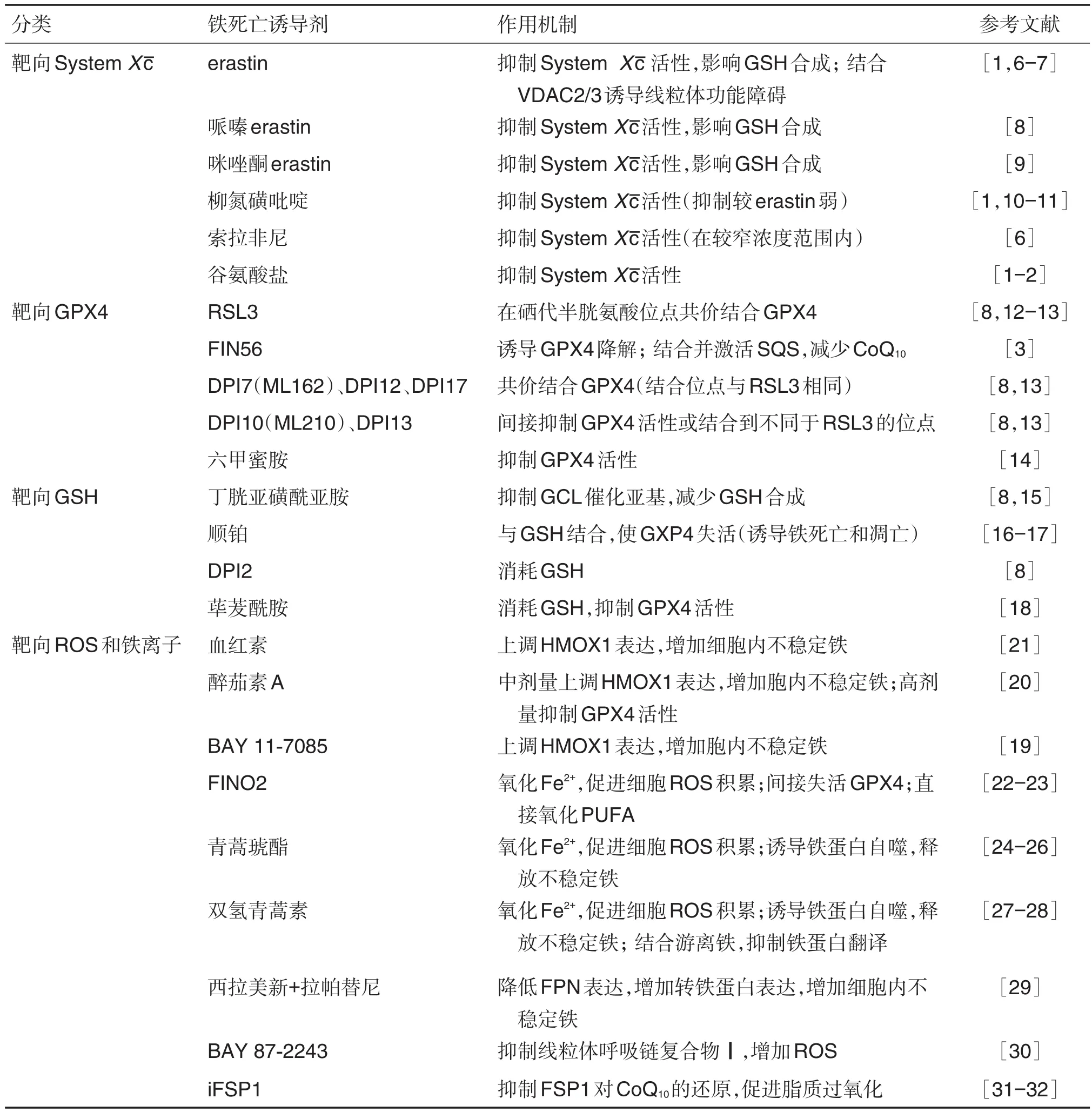
表1 铁死亡诱导剂及其作用机制
erastin水溶性差且代谢不稳定的特点限制了其体内应用,将哌嗪基引入erastin得到一种名为哌嗪erastin(piperazine erastin,PE,图2-2)的衍生物,其诱导HT-1080细胞铁死亡的作用机制与erastin相似,但水溶性和代谢稳定性优于erastin[8]。另一种衍生物咪唑酮erastin(imidazole ketone erastin,IKE,图2-3)具有纳摩尔级效价、高代谢稳定性和中等水溶性,可抑制小鼠弥漫性大B细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL)的生长;在DLBCL异种移植模型中,使用聚乙二醇-聚乳酸-羟基乙酸共聚物纳米粒载体可增加IKE在肿瘤中的蓄积,与游离IKE相比毒性更小,治疗指数更高[9]。
1.1.2 柳氮磺吡啶(sulfasalazine,SAS)
SAS是FDA批准的用于类风湿关节炎的一线抗炎药物。在体外,SAS可特异性抑制SystemXc,明显抑制淋巴瘤细胞增殖。大鼠腹腔注射SAS对宿主无明显损伤,但能显著减慢移植Nb2-U17淋巴瘤的生长,表明该药物在淋巴母细胞瘤的治疗中具有潜在的应用前景[10]。SAS在BJ细胞系(BJeH,BJeHLT,BJeLR和DRD)中的RAS选择性致死效力远低于erastin[1]。最近研究发现,CISD2的表达与SAS耐药有关,沉默Cisd2基因可通过增加线粒体Fe2+和脂质ROS的蓄积,使耐药的头颈癌细胞对SAS敏感而诱发铁死亡[11]。
1.1.3 索拉非尼(sorafenib)
索拉非尼以前称为BAY43-9006,是一种多激酶抑制剂,临床上用于治疗晚期癌症(如肾细胞癌、肝细胞癌和甲状腺癌)。索拉非尼对SystemXc的抑制具有浓度依赖性,仅在较窄的浓度范围内表现诱导HT-1080细胞铁死亡的作用。对87种索拉非尼类似物诱导HT-1080细胞铁死亡活性的分析表明,索拉非尼抑制SystemXc活性的机制可能是通过抑制一种未知激酶(其活性是SystemXc活性必需的)或与非激酶靶点(含有与索拉非尼激酶活性位点相似的结合口袋)相互作用[6]。
1.1.4 谷胺酸盐
高浓度谷氨酸盐也可以抑制SystemXc活性而诱导脑细胞和癌细胞发生铁死亡,这是生理条件下铁死亡的自然触发因素,可部分解释蓄积的谷氨酸盐对神经系统的毒性作用[1-2]。
1.2 靶向GPX4
GPX4具有维持细胞内GSH的动态循环和清除过氧化物的能力,是铁死亡的中枢调节因子。在细胞内半胱氨酸和GSH水平正常的情况下,也可以通过直接抑制GPX4活性诱导铁死亡。
1.2.1 RAS合成致死分子RSL3
RSL3 是 STOCKWELL 等[12]在筛选对致癌Ras合成致死小分子时发现的。后来通过基于亲和性的化学蛋白质组学,GPX4被鉴定为(1S,3R)-RSL3(图2-4)的靶蛋白。(1S,3R)-RSL3直接与GPX4共价结合,其活性基团是氯乙酰胺部分[8],主要通过烷基化硒代半胱氨酸(GPX4的活性位点)导致GPX4不可逆性失活,诱导铁死亡[13]。在RSL3的4种非对映异构体中,(1S,3R)-RSL3对BJ衍生细胞系的选择性杀伤力是其他3种异构体的100倍[8]。STOCKWELL等[12]发现RSL3的同时也发现了RSL5,并通过shRNA沉默HT-1080细胞中的Vdac3基因,证明RSL5与erastin一样,通过靶向VDAC发挥诱导铁死亡的作用。
1.2.2 衍生于ClL56的铁死亡诱导剂FlN56
SHIMADA 等[3]在 HT-1080 和工程转化的BJeLR细胞中检测了3169种诱导胱天蛋白酶非依赖性致死化合物,发现451种化合物在不激活胱天蛋白酶3/7的情况下触发细胞死亡,其中CIL56可能参与铁死亡和坏死2种细胞死亡途径。在CIL56的结构中,肟基是诱导铁死亡的必需基团,且哌啶部分的疏水性与效价相关。将CIL56的2个哌啶基替换为2个环己胺,得到在BJ细胞系中表现出更强抑制Ras突变癌细胞活性的药物FIN56(图2-5)。FIN56可以依赖乙酰辅酶A羧化酶,诱导GPX4蛋白翻译后降解导致细胞铁死亡。化学蛋白质组学分析还发现,FIN56可以结合并激活角鲨烯合酶(squalene synthase,SQS),导致辅酶Q10(coenzyme Q10,CoQ10)缺失,增强细胞对FIN56诱导铁死亡的敏感性。此外,化合物DPI7(ML162),DPI10(ML210),DPI12,DPI13和DPI17等处理的BJeLR细胞也缺乏 GPX4 活性[8],其中 DPI7,DPI12 和DPI17具有与RSL3相同的结合位点,而DPI10和DPI13间接抑制GPX4或结合到与RSL3不同的位点[13]。通过生物信息学分析,发现一种美国FDA批准用于卵巢癌治疗的抗肿瘤药物——六甲蜜胺(altretamine)可抑制GPX4的脂修复活性,提示其抗肿瘤活性的潜在机制[14]。
1.3 靶向谷胱甘肽
进入细胞内的胱氨酸转化成半胱氨酸,与谷氨酸通过谷氨酸-半胱氨酸连接酶(glutamate cysteine ligase,GCL)催化生成γ-谷氨酰半胱氨酸,随之在谷胱甘肽合成酶的作用下,与甘氨酸生成GSH。GSH是GPX4酶活性的重要辅助因子。
1.3.1 丁胱亚磺酰亚胺(buthionine sulfoximine,BSO)
BSO是GSH合成限速酶GCL的不可逆抑制剂,可以抑制GCL催化亚基,降低GSH水平和GPX4活性,诱导脂质过氧化,导致人胰腺癌细胞PANC-1、人结肠癌细胞HT-29和BJeLR细胞发生铁死亡。BSO与铁合用,可以增强抗癌效果[8,15]。
1.3.2 顺铂
顺铂等铂类化合物与富含巯基的生物分子具有高亲和力,可与GSH直接结合形成Pt-GS复合物,引起GSH耗竭和GXP4失活。顺铂诱导的细胞死亡是铁死亡和凋亡的共同结果,与erastin联合作用于人肺癌细胞A549和人结肠癌细胞HCT116,表现出明显的协同抗肿瘤活性[16]。近期,针对骨肉瘤细胞的研究表明,长期使用顺铂可能通过过度激活信号转导与转录激活因子3(signal tranducers andactivators of transcription 3,STAT3)信号,增强Nrf2的表达和活性,导致细胞抗氧化能力增强,造成对顺铂诱导铁死亡的耐药。联合使用其他铁死亡诱导剂(如erastin和RSL3)或STAT3抑制剂,可以增强细胞对顺铂的敏感性,为耐药骨肉瘤的治疗提供了新思路[17]。
1.3.3 其他
DPI2能消耗BJeLR细胞中90%GSH,表明其可能通过与上述化合物类似的机制诱导铁死亡[8];荜茇酰胺(piperlongumine,PL)在不降低细胞内GPX水平的情况下,通过消耗GSH而诱导PANC-1细胞铁死亡[18]。
1.4 靶向铁离子和活性氧
1.4.1 HMOX1和血红素
Nrf2的靶基因表达产物包括HMOX1,SLC7A11和GPX4等。在氧化应激时,Nrf2会与KEAP1解离,转位进入细胞核,与抗氧化反应元件结合,激活目标抗氧化基因的表达[20]。HMOX1兼有细胞保护和促进铁死亡的作用,保护作用归因于其抗氧化活性,而其毒性作用是由于它能催化血红素降解为Fe2+、胆绿素和一氧化碳,大量游离Fe2+通过芬顿反应促进细胞铁死亡。HASSANNIA等[20]认为,HMOX1水平的上调程度可能决定它促进还是抑制铁死亡。NAVEENKUMAR等[21]则发现,抑制铁死亡会显著减弱血红素诱导的血小板活化。血红素能通过上调HMOX1表达,增加细胞中不稳定铁,并降低GSH水平,导致LPO生成增多,诱导血小板发生铁死亡。HMOX1的特异性抑制剂二氯化锡原卟啉Ⅸ可以保护血小板免于血红素介导的细胞毒性,几乎完全抑制脂质过氧化作用,控制细胞内ROS水平。
1.4.2 醉茄素A(withaferin,WA)
WA(图2-6)是从南非醉茄中分离的甾体内脂类物质,在神经母细胞瘤中具有双重铁死亡诱导机制,中剂量的WA靶向作用于KEAP1,激活Nrf2上调HMOX1的表达;高剂量的WA可直接使GPX4失活。这为高危神经母细胞瘤的治疗提供一种有效策略[20]。
1.4.3 BAY 11-7085
CHANG等[19]发现,NF-κB抑制剂BAY 11-7085〔(E)-3-(4-叔丁基苯磺酰基)-2-丙烯腈〕也能通过Nrf2-SLC7A11-HMOX1途径上调HMOX1和游离铁,以NF-κB非依赖性方式诱导癌细胞发生铁死亡。
1.4.4 FlNO2
FINO2是一类具有1,2-二氧戊烷结构的plakinic酸D衍生物,可诱导人急性淋巴白血病细胞RS4;11铁死亡[22]。FINO2通过发生类芬顿反应氧化Fe2+和间接失活GPX4的共同作用诱导HT-1080细胞铁死亡。同时由于高亲脂性,FINO2可能会蓄积在膜脂双层中,直接氧化多聚不饱和脂肪酸(polyunsaturated fatty acid,PUFA)。与其他含有过氧键的化合物不同,FINO2能优先启动铁死亡而非其他形式的细胞死亡[23]。由于FINO2的作用具有铁依赖性,FINO2在铁水平高的恶性细胞中作用更强[22]。构效关系研究表明,过氧键及其附近的极性羟基是FINO2的活性基团,且羟基与过氧键必须有特定的空间关系,这是结合活性铁所必需的,还可能有助于过氧键的还原。用非极性基团取代羟基或增大羟基与过氧键之间的距离都会降低FINO2的效力[23]。(-)-FINO2(图2-7)对癌性成纤维细胞BJeLR的活性和选择性比其对映体更高[22]。
1.4.5 青蒿素类
青蒿素及其衍生物如青蒿琥酯(artesunate,ART,图2-8)和双氢青蒿素(dihydroartemisinin,DHA,图2-9)等,是一组有抗疟疾作用的倍半萜内酯。此外,这些化合物也被证明有抑癌作用。ART在KRas转化的胰腺导管腺癌细胞中可作为铁死亡特异性诱导剂,该细胞由于KRas的重新编程,对凋亡具有高度抵抗性,但可被ART诱导发生铁死亡,以溶酶体形式供给外源性铁可进一步增强ART的铁死亡诱导作用[24]。ART还可通过调节肝星状细胞中铁蛋白自噬介导铁死亡而减轻肝纤维化[25]。另一项研究也发现,ART可依赖P53诱导铁死亡,防止四氯化碳诱导的小鼠肝损伤和纤维化,并抑制肝星状细胞活化[26]。
与FINO2类似,DHA结构中的过氧键也可被Fe2+裂解并生成毒性自由基,促进细胞ROS的积累。DHA还可通过激活AMPK磷酸化,下调mTOR/p70S6k信号通路,加速铁蛋白通过自噬降解而增加铁池中铁的含量,有效诱导急性髓系白血病细胞铁死亡,并伴有线粒体功能紊乱[27]。另一项研究则发现,DHA通过与细胞内游离铁结合〔结合的铁具有与游离铁相同的氧化活性,但不能被铁调节蛋白-铁应答元件(iron regulatory protein-iron response element,IRP-IRE)铁稳态调节机制所识别〕而刺激IRP与含IRE序列的mRNA结合,抑制铁蛋白翻译,同时诱导铁蛋白溶酶体降解,增加细胞游离铁水平,进而增加细胞对铁死亡的敏感性[28]。
1.4.6 西拉美新(siramesine)和拉帕替尼(lapatinib)
溶酶体破坏剂西拉美新和酪氨酸激酶抑制剂拉帕替尼合用,可通过降低铁转运蛋白(ferroportin,FPN)的表达和增加TF的表达,导致细胞内铁离子蓄积,诱导MDA MB 231,MCF-7,ZR-75和SKBr3乳腺癌细胞发生铁死亡,这为乳腺癌的治疗提供新的策略[29]。
1.4.7 BAY 87-2243
BAY 87-2243可抑制线粒体呼吸链复合物Ⅰ,刺激线粒体膜通透性转换孔(mitochondrial permeablity transition pore,mPTP)开放和线粒体膜去极化,导致自噬体形成,触发线粒体自噬依赖的ROS增加,促进脂质过氧化和GSH消耗,导致BRAFV600E黑色素瘤细胞坏死样凋亡和铁死亡[30]。
1.4.8 iFSP1
最近,DOLL等[31]和BERSUKER等[32]同时发现了铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)通过还原CoQ10而阻止脂质过氧化,抑制铁死亡的发生。DOLL等还通过在药物筛选,发现FSP1的抑制剂iFSP1(图2-10)可在过表达FSP1的Gpx4敲除Pfal和HT-1080细胞中选择性诱导铁死亡。
2 铁死亡抑制剂
铁离子的蓄积和LPO生成增多是铁死亡的重要标志,脂质过氧化可通过酶促(含铁酶介导)和非酶促(芬顿反应)途径发生。目前已有的铁死亡抑制剂(图3,表2)主要通过消除自由基、抑制产生脂质或脂质过氧化物的酶、减少游离铁等途径发挥作用。
2.1 芳胺类抗氧化剂
ferrostatin-1(Fer-1,图3-1)是一种通过小分子文库高通量筛选得到的铁死亡特异性抑制剂,能够防止erastin或RSL诱导的脂质ROS蓄积,抑制HT-1080细胞铁死亡,但不抑制其它氧化致死化合物(如H2O2和鱼藤酮)和凋亡诱导剂(如氰化羰基-3氯苯腙)诱导的细胞死亡[1]。此外,Fer-1还可以下调前列腺素内过氧化物合成酶2表达及上调GPX4和Nrf2蛋白表达,通过抑制氧化应激,降低ROS和脂质过氧化作用,保护HT-22细胞免受谷氨酸诱导的氧化毒性和铁死亡[33]。在BEAS-2B细胞和炎性肺损伤小鼠模型中,Fer-1则通过抑制脂质过氧化,防止脂多糖诱导的铁死亡[34]。
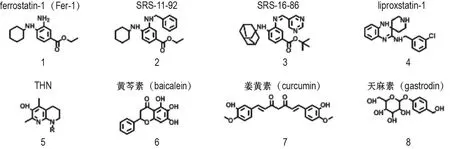
图3 部分铁死亡抑制剂的化学结构.THN:四氢萘啶醇;TEMPO:2,2,6,6-四甲基哌啶氧化物;PHOXNO:吩噁嗪氮氧化物.

表2 铁死亡抑制剂及其作用机制
分子构效关系表明,用硝基取代芳伯氨基或消除环己胺基均会破坏其清除自由基和抑制铁死亡的能力[35]。其中,环己胺基部分可能通过亲脂性使Fer-1能够锚定在脂膜上,而不影响分子固有的抗氧化能力。增加环己胺基部分的亲脂性可增加其铁死亡抑制活性[1],而将杂原子引入该部分则会导致效价降低[35]。
SKOUTA等[35]研究发现,Fer-1可能通过其互变异构体释放2个质子和2个电子,形成氧化还原稳定的化合物,作为还原剂发挥抗氧化活性。若2个氮原子上取代有阻止互变异构的基团,则活性会丧失。他们还发现一些比Fer-1更有效的第二代ferrostatin类化合物,如 SRS-11-92(图 3-2,EC50=6 nmol·L-1)的活性比 Fer-1(EC50=95 nmol·L-1)强15倍。LINKERMANN等[36]则发现了在erastin处理的HT-1080和NIH3T3细胞中可发挥铁死亡抑制作用的第三代ferrostatin类化合物SRS-16-86(图3-3),与Fer-1相比有更高的代谢和血浆稳定性,即使在急性肾损伤异常严重的情况下,也能发挥强大的保护作用。
MIOTTO等[37]针对Fer-1的作用机制进行研究,发现Fer-1可清除烷氧自由基而自身不被消耗,并与Fe2+络合减少细胞中不稳定铁,提出了一种Fer-1还原烷氧自由基而Fe2+还原Fer-1自由基的循环作用机制。
liproxstatin-1(Lip-1,图3-4)是在他莫昔芬诱导的Gpx4-/-小鼠胚胎成纤维细胞中,筛选4万多个类药小分子发现的抑制铁死亡的螺喹喔啉胺类化合物。Lip-1是LPO的特异性抑制剂,可在低纳摩尔范围内抑制铁死亡且不干扰其他经典类型的细胞死亡(如TNF-α诱导的凋亡和H2O2诱导的坏死等)[38]。LI等[39]发现,Lip-1可通过激活Nrf2信号通路,降低ROS,下调TGF-β1,减轻放射性肺纤维化,为放射性肺纤维化提供了一个新的治疗靶点。而FENG等[40]则发现,Lip-1可在不影响触发mPTP开放所需Ca2+量的情况下,通过下调VDAC1(不影响VDAC2/3)水平和寡聚化,减少线粒体ROS产生,恢复GPX4水平,缩小离体小鼠缺血/再灌注心肌的梗死面积,发挥心脏保护作用。
最近,SHENG等[41]对Lip-1灭活LPO的机制进行了详细研究,发现Lip-1与Fer-1具有类似的作用机制。Lip-1易渗透到脂双层并停留在其内部,使其活性位点与脂质过氧化位点紧密定向接触,启动CH3OO·从芳香胺位提取氢原子。清除LPO后,Lip-1形成的Lip-1自由基可被体内其他抗氧化剂(如泛醌)还原。他们还对liproxstatin类化合物进行了构效关系研究,发现芳胺结构是这类铁死亡抑制剂抗氧化活性所必需的。
尽管在溶液中,Fer-1和Lip-1对自由基的反应性比α-生育酚低,但由于它们具有弱氢键相互作用和更优的体内动力学特征,能在脂质双分子层中发挥比α-生育酚更好的抑制作用[42]。
2.2 α-生育酚类似物
α-生育酚主要通过破坏自动氧化的链式反应发挥抗氧化能力。其衍生物四氢萘啶醇(tetrahydronaphthyridinols,THN,图3-5)是一种特殊的氮杂酚类,由酚类抗氧化剂芳香环上相对羟基的3和(或)5位引入氮原子而来。在(1S,3R)-RSL3诱导的Pfa-1小鼠胚胎成纤维细胞中,亲脂性THN与过氧自由基的反应速度几乎是α-生育酚的30倍,且含有12~15个碳烷基链的THN具有最佳的抑制效果,甚至比Fer-1和Lip-1的活性更好[42]。
2.3 氮氧化物
具有细胞膜渗透性并能穿过血脑屏障的2,2,6,6-四甲基哌啶氧化物(TEMPO)能将柠檬酸亚铁氧化,阻断芬顿反应,抑制羟基自由基的产生[43]。此外,单芳基氮氧化物、二芳基氮氧化物和吩噁嗪氮氧化物等,都被认为是极具前景的脂质过氧化和铁死亡的强效抑制剂。其中吩噁嗪氮氧化物(PHOXNO)抑制RSL3诱导的小鼠胚胎成纤维细胞铁死亡最有效,且效果明显优于Fer-1和Lip-1[44]。
2.4 多酚类天然化合物
最近,越来越多的天然产物被发现具有抑制铁死亡的作用,它们大多为多酚类物质,可能通过多种途径发挥铁死亡抑制作用。
XIE等[45]在天然产物库筛选铁死亡抑制剂的实验中发现,黄芩素(baicalein)(图3-6)可能通过抑制GSH耗竭、GPX4降解以及脂质过氧化而抑制erastin诱导的胰腺癌细胞铁死亡,还可能激活Nrf2通路,阻止erastin诱导的Nrf2降解,抑制氧化损伤。此外,黄芩素可通过抑制12/15-脂氧合酶(12/15-lipoxygenases,12/15-LOX),减少肝缺血再灌注后的细胞死亡[46],且能在柠檬酸铁铵诱导的HT-22细胞损伤模型和FeCl3诱导的小鼠癫痫发作中发挥神经保护作用[47]。最新研究表明,黄芩素还能降低铁死亡中磷脂酰乙醇胺氧化,改善脑皮质撞击的预后恢复[48]。
此外,姜黄素(curcumin,图3-7)则可能作为铁螯合剂减少铁蓄积,防止erastin诱导的小鼠胰岛细胞瘤胰腺β细胞铁死亡[49],还在横纹肌溶解诱导的急性肾损伤小鼠模型中被证明具有铁死亡抑制作用,并可增强HMOX1活性[50]。天麻素(gastrodin,GAS,图3-8)可通过Nrf2-HMOX1信号通路,诱导GPX4,FPN1和ACSL4等表达,抑制谷氨酸诱导的HT-22细胞铁死亡[51]。
2.5 酯酰辅酶A合成酶长链家族成员4抑制剂
噻唑烷二酮类药物(thiazolidinediones,TZD)是已上市的胰岛素增敏剂,可激动过氧化物酶体增殖物激活受体γ(peroxisome proliferator activatedreceptor γ,PPARγ)。酯酰辅酶A合成酶长链家族成员4(acyl-CoA synthetase long-chain family member 4,ACSL4)专司游离脂肪酸的酯化,在合成长链PUFA-CoA中起关键作用。研究发现,罗格列酮、吡格列酮和曲格列酮等可特异性抑制ACSL4(不抑制其他ACSL亚型),预防RSL3诱导的Pfa1细胞铁死亡和脂质过氧化,其机制与PPARγ信号通路无关。其中,曲格列酮虽抑制ACSL4的效力较低,但可能由于其6-色原烷醇结构而具有固有抗氧化活性,是TZD中最具保护性的[52]。
2.6 脂氧合酶抑制剂
齐留通(zileuton)是5-LOX的口服特异性抑制剂,用于哮喘患者的维持治疗。它能通过抑制5-羟基二十烷四烯酸的生成,保护ACSL4过表达的LNCaP和K562细胞免于erastin诱导的铁死亡[53]。
2.7 其他
铁螯合剂去铁胺(deferoxamine)、心脏保护药右雷佐生(右丙亚胺,dexrazoxane)等也具有抑制铁死亡的作用。
3 结语
铁死亡是调节性坏死的一种重要形式,在形态学、生物化学和遗传学上具有不同于其他细胞坏死和凋亡的特征。铁死亡的发生机制与细胞代谢密切相关,涉及多种关键分子和信号通路,调控这些关键分子合成或分解及其涉及的信号通路,会改变细胞对铁死亡的敏感性。合理诱导或抑制细胞铁死亡,将有助于改善和治疗特别是癌症和损伤相关的多种疾病。
目前已发现各种类型的铁死亡诱导剂和抑制剂,然而对其中大多数化合物的作用靶点和潜在应用仍有待深入了解。进一步阐明这些化合物(尤其是多作用靶点化合物)的作用机制、各机制间的关联和作用特点,同时探讨联合用药的可能性以及开发特异性更高的诱导剂或抑制剂,都将对其临床应用产生深远影响。
