Molecular Structure of Two Glutamate Decarboxylases From Mung Bean[Vigna Radiate (L.)] Analyzed by Spectroscopy
2020-12-04WANGXianqingWEITongYANGYongSHIYanguo
WANG Xian-qing, WEI Tong, YANG Yong, SHI Yan-guo
1. College of Food Science, Heilongjiang Bayi Agricultural University, Daqing 163319,China 2. Beijing Shunxin Holdings, Beijing 101300, China 3. College of Food Science, Harbin University of Commrce, Harbin 150076,China
Abstract Glutamate decarboxylase (GAD) catalyzes an α-decarboxylation reaction of glutamate to produce γ-aminobutyric acid (GABA). Interestingly, two kinds of mung bean [Vigna radiate (L.)] GADs were firstly extracted and purified in this research. Two GADs in mung bean were both dimers with a molecular mass of 155 kDa (GAD1) and 75 kDa (GAD2). The enzymatic properties of GAD1 and GAD2 were detected in this research. Infrared spectroscopy analysis revealed that more higher-ordered structure contents, α-helix and β-sheet structures, was found in GAD2, which determined the higher stability of GAD2. It was analyzed by Raman that the molecular structures of GAD1 and GAD2 are generally “exposed”. Fluorescence analysis revealed that GAD1 had a more flexible and exposed molecular structure, while the conformation of GAD2 was more compact and conservative. It was found that pH and temperature-induced structural change decreased the enzyme activity. Ca2+ was involved in binding the calmodulin-binding domain of GADs and induced a “buried” and compact structure. The unfolding of GAD induced by SDS, impaired the enzyme activity. KI, MgSO4, AgNO3, and SDS significantly inhibited GAD1 and GAD2 activities. Tween 80, Ca2+ and Cu2+ could significantly activate GAD1 and GAD2, and Fe2+ only increased GAD2 activity.
Keywords Glutamate decarboxylase; Mung bean; Structure; Spectroscopy
Introduction
Glutamate decarboxylase (GAD) catalyzes the α-decarboxylation reaction of glutamate to produce γ-aminobutyric acid (GABA). GAD has been shown to play an important role in the regulation of brain excitability through the synthesis of GABA and it is considered a specific marker for GABAergic neurons and their processes[1]. As both GABA, and GAD are widely distributed in living cells of various organisms from single-cell organisms to mammals, the role of GAD has been explored outside the mammalian neuronal system[2]including being used as an antigen for the diagnosis and prediction of insulin-dependent diabetes mellitus (IDDM); purified and characterized from rice bran[2], potato[3], and wheat[4]. It is speculated that plant GAD has species-specific functions[5]. GAD activity is also affected by stress-related conditions, including anaerobiosis, cytosolic pH reduction, cold-stress, and heat-stress.
Mung bean [Vigna radiate (L.)] has been eaten since ancient times. It has been traditionally grown in most parts of China and has been accepted as part of the local diet in soups and desserts[6]. Mung bean is a popular food in China due to its detoxifying, anti-inflammatory, antitumorigenic, cholesterol-lowering, and diuretic properties[7]. Previous research is focused on GAD application on GABA production, and to the best of our knowledge, there is no information available on the purification and characterization of the mung bean GAD. Understanding of the physicochemical properties of mung bean’s GAD may lead to the production of GABA enriched foods. Therefore, in this paper, we report the purification and physicochemical properties of mung bean’s GAD.
1 Materials and methods
1.1 Materials
Mung beans were obtained from the local market within three months after harvest and were stored at room temperature during the studies. Analytical grade chemicals and reagents were purchased from Sigma Chemical Corp. (MO, USA).
1.2 Determination of GAD activity
The reaction mixture consisted of 200 μL of 50 mmol·L-1sodium phosphate, pH 5.6, 100 mmol·L-1L-glutamate, 0.2 mmol·L-1Pyridoxal phosphate (PLP), and 100 μL of the enzyme. The reaction solution was incubated at 40 ℃ for 60 min, and then the reaction was terminated by the addition of 100 μL of 32% (W/V) trichloroacetic acid (TCA). The suspension was filtered through a 0.45 μm membrane filter (Whatman, USA). The filtrate was analyzed for GABA content using Agilent’s 1100 HPLC (Agilent Technologies, USA). One GAD activity unit was defined as the release of 1 μmol of GABA produced from L-glutamate per 30 min at 40 ℃. Specific activity was defined as GAD activity units per mg of the enzyme[8].
1.3 Preparation and purification of GAD
Two hundred grams of mung beans were homogenized at 10 000 r·min-1for 15 min in 1 L of 50 mmol·L-1sodium phosphate buffer, pH 5.6, containing 0.2 mmol·L-1PLP, and 2 mmol·L-12-mercaptoethanol (ME), 2 mmol·L-1Ethylenediaminetetraacetic acid (EDTA), and 1 mmol·L-1Phenylmethanesulfonyl fluoride (PMSF). After homogenization, the protein extract was filtered through four layers of cheesecloth and centrifuged at 7 000 g for 20 min at 4 ℃. Solid ammonium sulfate was then added to this crude extract up to 30% saturation and centrifuged at 10 000 g for 20 min at 4 ℃. The supernatant was adjusted to 50% saturation by further addition of solid ammonium sulfate and centrifuged again at 10 000 g for 20 min at 4 ℃. The precipitate was dissolved in 100 mL of the standard buffer. The mixture was dialyzed three times against 2 L of the standard buffer. The supernatant of the dialysate was called the crude GAD and used for further purification. Protein concentrations in various preparations were assayed by Bradford’s method.
Crude GAD was first purified by a 2.5×25 cm DEAE-Sepharose FF ion exchange column which was eluted with a linear gradient of 0~1.4 mol·L-1NaCl in the standard buffer. The enzyme activity peak was obtained for further purifying. The dialyzed enzyme purified by gel filtration chromatography with a 1.0×100 cm Superdex-200 column which was pre-equilibrated with the standard buffer. Two fractions with high GAD activities were collected again. Two GADs separated were loaded on affinity chromatography with Glu-Sepharose CL 4B column that eluted with 300 mL of a linear gradient of 0~0.5 mol·L-1NaCl in the standard buffer[9].
1.4 Determination of subunit composition of GAD by SDS-PAGE profile
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed, according to Liu et al.[10]. GAD was mixed with two volumes of loading buffer. Aliquots of 5 μL of each sample were loaded on a 5% stacking gel and a 10% polyacrylamide resolving gel with a constant voltage of 120 V. Then the gels were stained with Coomassie Brilliant Blue R250 for protein visualization and scanned using a Gel DocTM EZ Imager (BIO-RAD).
1.5 Determination of molecular weight of GAD by analytical gel filtration
The purified GAD was loaded on a Sephacryl S-100 HR 16/60 column equilibrated with phosphate buffer (PB) containing 0.15 mol·L-1NaCl and EDTA, and eluted with the same buffer at a flow rate of 0.5 mL·min-1. Amylase (200 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.4 kDa) were used as molecular weight standards. All standards were purchased from Sigma-Aldrich (St. Louis, MO).
1.6 Effect of pH on GAD activity
The optimum pH for GAD activity was determined within a range of pH 3.5 to 8.5 in 0.5 mol·L-1citrate or sodium phosphate buffer supplemented with 50 mmol·L-1glutamate and 10 mmol·L-1PLP at 37 ℃. The pH stability of the enzyme was determined by incubating it at each pH (4, 5, 6, 7, and 8) with 0.5 mol·L-1citrate or sodium phosphate buffer for 1 h at 4 ℃, followed by residual activity measurement.
1.7 Effect of temperature on GAD activity
The optimum temperature for GAD activity was determined during a 30 min exposure to a temperature range of 20 to 60 ℃ in 0.5 mol·L-1citrate buffer (pH 5.0) with 50 mmol·L-1of glutamate and 10 mmol·L-1of PLP. The thermal stability of the enzyme was determined by incubating the enzyme in a temperature range of 20 to 60 ℃ for 1 h at pH 5.0, and the residual activity of the enzyme was measured at 37 ℃.
1.8 Effect of various reagents on GAD activity
Fifty microliters (2 mmol·L-1) of either KCl, KI, MgSO4, MnSO4, Al2(SO4)3, AgNO3, Li2SO4, HgCl2, or CaCl2were incubated with 50 μL of the enzyme (0.2 mg·mL-1) at 30 ℃ for 30 min. The remaining GAD activity was measured as described above.
1.9 Fourier transform infrared spectroscopy (FTIR) analysis
FTIR absorption spectra from 4 000 to 400 cm-1were acquired in the transmission mode by a Nicolet Magna IR 550 FTIR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) continuously purged with dry air and equipped with a liquid nitrogen cooling MCT detector. GAD samples were first freeze-dried and then produced by pressing in KBr windows (1.5 mg protein to 200 mg KBr) on a Carver press at 5~6 T pressure. Each spectrum was obtained by co-adding 256 interferograms at a spectral resolution of 2.0 cm-1. The decomposition of the amide Ⅰ band was performed in the region of 1 700~1 600 cm-1. The quantitative analysis of the secondary structural components of GAD was determined by a Peak fitting software according to the method of Wang’s[11].
1.10 Raman spectroscopic analysis
To perform a Raman experiment, 150 mg of GADs are considered. Raman spectra are recorded on a Perkin Elmer Raman Station 400F Dispersive Raman Spectrometer, that is equipped with a 785 nm diode laser, which is used as depending on the fluorescence contribution of the sample. The laser is focused on the samples, which are placed on microscope slides. Each spectrum is obtained under the following conditions: 80 mW of laser power; 4 scans; 60 s exposure time; 2 cm-1resolution; and the range of Raman spectra is about 400~2 000 cm-1. Each sample is scanned at least thrice, and the Raman spectra for each sample are plotted by calculating the mean. Errors in band position are less than ± 3 cm-1.
1.11 Fluorescence spectroscopy analysis
Fluorescence spectroscopy of mung bean’s GADs was measured according to the methods of Zhang et al.[12]. The enzyme samples with or without the addition of chemical reagents were analyzed in 50 mM phosphate buffer at pH 4.0, 5.5, and 7.0 on a FluoroMax-4 fluorescence spectrometer (HORIBA Scientific, American) at 25 ℃ and 40 ℃. The temperature was controlled by pumping warm water into a thermal insulation device kept outside of the measuring cup. GAD solutions (0.03 mg·mL-1) were excited at 290 nm, and the emission spectra were recorded from 300 nm to 450 nm.
1.12 Statistical analysis
The experiments were performed in triplicate, and Student’s t-test and ANOVA with the post-hoc test were performed, when appropriate, to evaluate means and standard deviation. A maximum of 5% standard deviation from the averaged values was generally tolerated (when not otherwise specified). The averaged values are documented in the respective figures.
2 Result and discussion
2.1 Molecumar composition of GAD
GAD in mung bean was purified by DEAE-Sepharose FF ion-exchange chromatography, Superdex-200 gel filtration chromatography, and Glu-Sepharose CL 4B affinity chromatography in this research. As depicted in Fig1A, it was interesting that there was one enzyme activity peak was observed in DEAE-Sepharose FF ion-exchange chromatography with a 57.2% recovery ratio, while we found two distinct peaks corresponding to two enzymes in Superdex-200 gel filtration chromatography [as shown Fig.1(b)]. The GAD with a higher molecular mass was labeled as GAD1 and the enzyme with less molecular mass was labeled as GAD2. The recovery ratio of Superdex-200 gel filtration chromatography for GAD1 and GAD2 were 23.1% and 19.7% respectively. It could be seen from Fig.2(a) and (b) that The further purification of GAD1 and GAD3 were performed by Glu-Sepharose CL 4B affinity chromatography with recovery ratios of 10.7% and 14.2%. As shown in Fig.3, we determined by analytical gel filtration the molecular mass for two kinds of GADs in mung bean, GAD1 has a molecular mass of 155 kDa and GAD2 has an apparent molecular mass of 75 kDa.

Fig.1 Purification of mung bean’s GAD by DEAE-Sepharose FF chromatography (a) and Superdex 200 chromatography (b)

Fig.2 Purification of mung bean’s GAD bySuperdex 200 chromatography(a): GAD1; (b): GAD2

Fig.3 HPLC separation of GAD1 and GAD2 from Mung Bean
In order to determine the subunits composition of GAD, SDS-PAGE analysis was performed as depicted in Fig.4. The SDS-PAGE profile of GAD1 showed a distinct subunit with a molecular weight of approximately 75 kDa, whereas for GAD2 a subunit with a molecular weight of 36 kDa was detected. It can be deduced that the two GADs identified in mung bean were dimers. Mung bean GAD2 is similar in molecular weight to rice germ GAD, which has a molecular weight of 40 kDa[2]. GADs extracted from other plants and microorganisms have been found to have different molecular weights. It was reported that the molecular weight of purified GAD from Aspergillus oryzae determined by SDS-PAGE and gel filtration was estimated to be 48 kDa and 300 kDa, respectively, suggesting that purified GAD had a hexameric structure[13]. Squash GAD was reported to be composed of multiple identical subunits with a molecular weight of 58 kDa[14]. In contrast, the molecular weight of the mushroom GAD was estimated to be 30 kDa under native conditions[15].
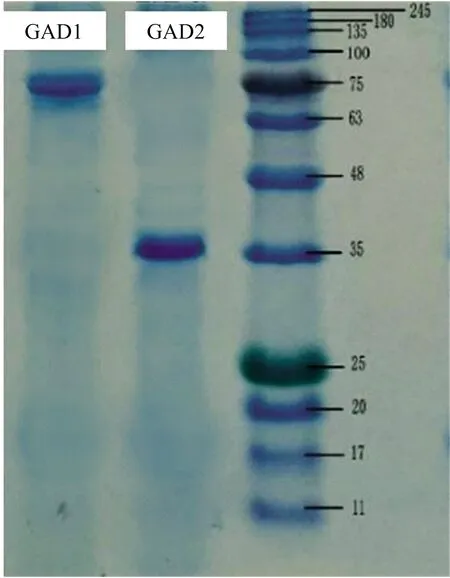
Fig.4 Subunit composition of mung bean GAD
2.2 Effect of pH and temperature on mung bean GAD activity
The pH-dependent stability and activity of GAD were examined, and the results are shown in Fig.5. The findings revealed the optimum activity for GAD1 and GAD2 were at pH 6.1 and pH 5.5, respectively. In comparison, the optimum pH of most plant GAD was reported to be between 5.5 and 6.5[16], bacterial GADs were low, and those of animal GAD have high optimum pH. The specific activity of mung bean GAD1 was higher than that of GAD2. Mung bean GAD1 was stable within a pH range of 5.3 to 5.9 but was unstable below pH 5.0 and above pH 6.5. While GAD2 was stable within a broader pH range (4.5 to 6.1), but the remaining enzyme activity decreased to 30% above pH 8.0. Therefore, GAD2 could be a potential candidate for applications in industry requiring broader pH stability ranges. The difference in pH stability between the two GADs might be related to the dissociation of the subunits. It was concluded in a previous study that plant GADs seemed to require an acidic pH for optimal activity[17]. Squash GAD was reported to be a hexamer which dissociates to a dimer, that has enzyme activity but appears to be less stable, at the unstable pHs[18]. Gut et al.[19]found that plant GAD was regulated by pH conditions and revealed a common structural basis for pH-mediated regulation where the β-hairpin in GAD acts as a pH-dependent switch, allowing plant GAD to respond flexibly to different kinds of cellular stress occurring at different pH values.
The activity of mung bean’s GAD1 and GAD2 were both optimal at 40 ℃, as depicted in Fig.6. The optimum temperatures for potato extracted GAD was reported to be 37 ℃[20], while squash GAD has a higher optimum temperature of 60 ℃[18]. The mung bean GAD1 was stable at temperatures of 30 ℃ and 45 ℃, while GAD2 was stable at a wider temperature range. The increase in stability of GAD2 in wider temperatures and pH ranges might be corresponding to its structure.

Fig.5 The optimum pH of pH stability of GAD activity(a): Optimum pH of GAD activity; (b): pH stability of GAD activity
2.3 Effect of chemical reagents on GAD activity
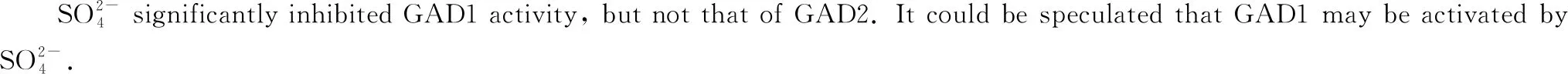
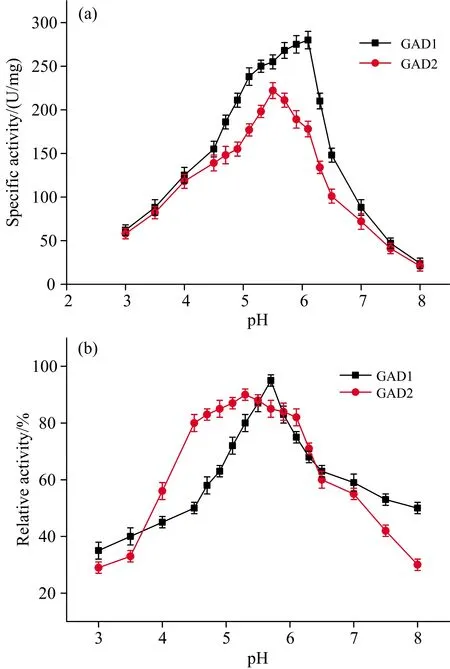
Fig.6 The optimum temperature of temperaturestability of GAD activity(a): The optimum temperature of GAD activity; (b): temperature stability of GAD activity

Table 1 Effect of chemical reagents on GAD activity
It was found that Tween 80 increased GAD1 and GAD2 activities, whereas SDS inhibited them. This could be due to an SDS-induced denaturation of GAD, which significantly decreases GAD1 and GAD2 activities. A similar inhibitory effect was observed in rice bran GAD, which showed an enzyme activity loss of 33.1%[17]. In comparison, the stability of GAD2 was higher than GAD1 when exposed to chemical reagents. Tween 80, a non-ionic surfactant, served as an emulsifier that could promote the enzymatic reaction.
Both Ca2+and Cu2+could significantly activate GAD1 and GAD2, whereas Fe2+only increased GAD2 activity. Plant GADs are be able to bind calmodulin (CaM) and require a CaM-Ca2+complex to enhance their enzymatic activity. Soy GAD was reported to be activatedviaCa2+signal transduction, which was stimulated 2- to 8-fold in the presence of calcium2+(Ca2+)/calmodulin (CaM) at natural pH[22]. The biotransformation activities of Bacillus megaterium significantly increased by Ca2+, Fe2+, and Cu2+[23]. No significant increase was observed in Fe2+stimulated GAD extracted from germinatedSetariaitalica(L.)Beauv[24].
2.4 Structure of GAD determined by FT-IR
The secondary structure of GAD1 and GAD2 were determined by FT-IR, as shown in Fig.7. The quantitative analysis of secondary structural components of proteins can be obtained by various experimental methods. Analysis of the second derivative of the IR-SD was considered as the most reliable quantitative method. A baseline was adjusted prior to the second-derivative analysis for the basis of further studying
of Fourier self-deconvolution, which then substantially affected the determination of the bands resolved by a Gaussian curve fit. The Gaussian curve fit was adjusted to give the best least-squares fit of the individual bands to each deconvoluted spectrum, followed by a second-derivative analysis[25]. In order to quantitatively analyze the contents of each secondary structures of GADs, a combination of Fourier self-deconvolution, second derivative analysis, and Gaussian curve fit was performed in this research.

Fig.7 FT-IR spectrum of GAD1 and GAD2

Table 2 Secondary structure of GAD1 and GAD2 determined by FT-IR
We found that GAD1 contained 35.35% α-helix structure, 19.76% β-sheet, 24.65% β-turn, and 22.24% unordered structure. While GAD2 had 37.89% α-helix structure, 24.88% β-sheet, 20.87% β-turn, and 16.36% unordered structure. In comparison, the higher-ordered structure contents determined the higher stability of GAD2. In comparison, it was observed that the GAD in rice germ had a higher β-sheet content of 39%[12]. It was reported that the brain GAD-cofactor PLP complex, holoenzyme (holoGAD), had an α-helix content of 34%, and a lower β-sheet content of 18% estimated by CD spectrum, while the brain apoGAD, whose cofactor PLP was removed, had a lower α-helix content of 24% and a higher β-sheet content[26]. The higher α-helix content in plant GAD might be related to the larger calmodulin-binding domain in the C-terminal segment of GAD is since this domain is likely to form an α-helical structure[27]. All plant GAD have been shown to have C-terminal CaM-binding domain, but bacterial and animal GADs lack of such a domain[28], this confirmed the higher α-helical content of GAD in plant versus in animals. GAD2 with a higher α-helix content might be inferred as holoGAD from the previous works, but it needed to be further confirmed.
2.5 Structure of GAD determined by Raman
The Raman spectra of the GAD1 and GAD2 are shown in Fig.8. The assignments of some major bands are based on our previous works[29]. The change in frequency and intensity of the Raman band mainly represents changes in the secondary structure and variations in the local environments of RBP.
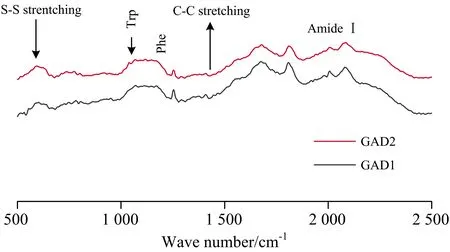
Fig.8 Raman spectrum of GAD1 and GAD2
The secondary structures of GADs were mainly determined by the Raman characteristic bands of the amide Ⅰ band. The Raman characteristic bands of amide I band are located as follows: α-helix, 1 645~1 660 cm-1; β-sheet, 1 670~1 680 cm-1; β-turn, 1 680~1 690 cm-1; random coil, 1 660~1 670 cm-1 [29]. The quantitative calculation of the secondary structures of the RBP Raman spectra was performed with Labspec software. The estimated percentage for the secondary structure of GAD1 and GAD2 were listed in Table 3. GAD1 contained 40.05% α-helix, 15.16% β-sheet, 15.65% β-turn, and 30.04% unordered structure, while GAD2 had 47.93% α-helix, 16.44% β-sheet, 10.83% β-turn, and 24.30% unordered structure. Though the contents of the secondary structure determined by Raman were different with those determined by FTIR, higher contents of α-helix and unordered structure in GAD1 were determined by both two methods.

Table 3 Secondary structure content of GAD1 and GAD2 determined by Raman

Table 4 Side chain group band intensity of GAD1 and GAD2
Tryptophan (Trp) residue showed several characteristic Raman bands, some of which are useful to check the polarity of the microenvironment, or participate in hydrogen bonding. Li-Chan[30]have reported that tryptophan residues from a buried hydrophobic microenvironment become exposed to the polar aqueous solvent, and there may be a decrease in the intensity of a band near 760 cm-1regions. As shown in Table 4, the Raman intensity of Trp of GAD2 was higher than that of GAD1, which suggested that GAD1 had a more “exposed” protein structure while GAD2 tend to “buried”.
The tyrosyl (Tyr) double ratio (I850/830) can be used in monitoring the microenvironment around the tyrosyl residues. In fact, when tyrosine residues were buried, then theI850/830ratio reached its minimum value of about 0.3, and the phenolic OH group acts as a strong hydrogen bond donor to an electronegative acceptor, such as carboxyl oxygen. When Tyr was exposed at the surface of the proteins, the phenolic OH acted as a donor and an acceptor for the moderate hydrogen bonds, and theI850/830was approximately 1.25[31]. It could be deduced that the Tyr of GAD1 and GAD2 tended to expose to the aqueous or polar microenvironment or act as simultaneous acceptor and donors of medium to weak hydrogen bonds. TheI850/830ratio of Tyr in GAD1 was significantly higher than GAD2, which suggested that more Tyr residues in GAD1 tend to “exposed”.
The band assigned to the CH2and CH3bending vibrations was observed around 1 450 cm-1. The decrease in the intensity of these bands indicates the exposure of aliphatic residues, while an increase indicates buried residues[32]. By monitoring the changes in the spectra around 1 450 cm-1(C—H2bending), the microenvironment of the aliphatic amino acid residues was investigated. It could be concluded that GAD1 had a higher Raman intensity of C—H2bending than GAD2, which suggested that more aliphatic amino acid residues in GAD1 tend to bury.
2.6 Structure of GAD determined by fluorescence spectrum
The tertiary structure of GAD was determined by fluorescence spectrum, as shown in Fig.9. GAD1 and GAD2 in pH 5.5 sodium phosphate buffer at 40 ℃ both exhibited one peak in their fluorescence emission spectra. The maximum emission (λmax) of GAD1 was located at 334.5 nm, whileλmaxof GAD2 was located at 333.1 nm. Commonly, when using Trp fluorescenceλmaxinformation, a Trp is assigned as being buried and in a “nonpolar” environment ifλmaxis <330 nm; ifλmaxis >330 nm, the Trp is assigned to be in a “polar” environment[33]. It could be deduced that the molecular structures of both GAD1 and GAD2, generally tend to “exposed” to a “polar” environment.
In comparison, GAD1 had the higher fluorescence intensity and largerλmax, which suggested that the microenvironment around the tryptophan residue of GAD1 tended to expose to more “polar” environment, resulting in a more flexible and exposed molecular structure, while the conformation of GAD2 was more compact and conservative, which corresponded to the higher enzyme stability.
Theλmaxof GAD1 in pH 4.0 buffer at 40 ℃ exhibited a red-shift from 334.5 nm (pH 5.5) to 337.1 nm, while theλmaxof GAD1 in pH 7.0 buffer tended to be increased up to 335.4 nm. This phenomenon proved that the inhibition of enzymatic activity might be related to the partial unfolding of the enzyme structure. An increasedλmaxfor GAD2 at pH 4.0 and 7.0 at 40 ℃ were also found when compared withλmaxat pH 5.5, which confirmed the pH-induced structural change decreased the enzymatic activity. The basis of pH-dependent absorbance changes was excluded that high pH induces the formation of an aldimine between the internal aldimine and a GAD cysteine residue and that the low pH conformation favors the unsubstituted internal aldimine[34]. The pH-dependent conformational change induces an alteration in the polarity of the active site affecting GAD activity[35].
Theλmaxof GAD1 significantly increased from 334.5 to 337.1 nm when the temperature increased from 40 to 60 ℃, whereas at room temperature decreased to 333.3 nm. Slight increases were observed inλmaxof GAD2 at 25 and 60 ℃, which implied that a decrease or increase in temperature causes a small conformational change in GAD2. The temperature stability of GAD2 was higher as a result of the slight structural change.
Some metal ions interfere with Fluorescence emission spectra. Thus only the fluorescence spectra of GAD combined with Ca2+and SDS were collected. The addition of Ca2+significantly decreased theλmaxof GAD1 and GAD2. This result confirmed that Ca2+was involved in the binding the calmodulin-binding domain of mung bean GAD then inducing a “buried” and compact structure of the enzyme. However, the addition of SDS significantly increased theλmaxof both GAD1 and GAD2, showing typical unfolding profiles, which indicated that the SDS-induced unfolding of GAD impaired the enzyme activity.

Fig.9 Fluorescence spectrum of GAD1 and GAD2
3 Conclusion
Glutamate decarboxylase (GAD) catalyzes an α-decarboxylation reaction of glutamate to produce γ-aminobutyric acid GABA. In this study, two GADs were extracted and purified from mung bean for the first time. It was interesting that, unlike GADs from other species, two purified GADs with different subunits composition, molecular structures, and enzymic properties were purified in this research. The two GADs identified in mung bean were both dimers.
The effect of pH, temperature, and chemical reagents on GAD enzyme activities were studied in this research. The inhibition and promotion effect of pH, temperature, and chemical reagents were partly revealed, we found that the GAD2 had strong structural stability, and structural change induced by pH and Ca2+, which may account for the changes in enzyme activities. In general, the higher-ordered structure contents determined the higher stability of GAD2. The GAD2 had a more conservative structure than GAD1 as depicted in the fluorescence spectrum. Additionally, GAD1 had a higher content of g-g-t conformation while GAD2 had t-g-t conformation.
Though some enzymatic properties and structural properties were determined in this research, further studies on the structure and regulation of mung bean GADs activity are needed. The amino acid sequence of GAD1 and GAD2 should be analyzed in further studies to provide more structural information for the function of GAD.
