铈锆固溶体CexZr1-xO2上H2S的选择性催化氧化性能
2020-12-01张凤莲蒋国霞郝郑平
孙 宇,张 鑫,张凤莲,蒋国霞,魏 征,郝郑平, *
(1.中国科学院生态环境研究中心环境纳米技术与健康效应重点实验室,北京 100085; 2.中国科学院大学环境材料与污染控制技术研究中心挥发性有机物污染控制材料与技术国家工程实验室,北京 101408)
硫化氢是石油化工、天然气行业生产过程中产生的一种重要的含硫化合物[1]。硫化氢对人体健康和环境都会造成很大的危害,在0.07% (物质的量分数)时即会造成人的死亡[2]。同时,其对管道和生产装置也具有极强的腐蚀性[3]。近年来,酸性气体排放的相关法律法规日益严格,《恶臭污染物排放标准》修订后,H2S的排放标准大幅度提高,这也对酸性气体的回收工艺提出了更高的要求。
Claus法是工业上应用最广泛的硫磺回收工艺。Claus工艺分为热反应阶段和催化反应阶段,在热反应阶段中,部分H2S被空气氧化为SO2;催化反应阶段中,反应炉内生成的SO2与H2S在催化剂的作用下生成单质硫[4],反应方程式如下:
(1)
(2)
反应(2)为可逆反应,受热力学平衡的限制,Claus尾气中仍有2%左右的H2S未被转化[2]。因此,Claus尾气的后处理技术至关重要。此外,很多小型工厂尾气中硫化氢的浓度较低,不适宜采用Claus尾气处理装置。而H2S选择氧化技术可以将硫化氢直接氧化成硫磺,不受热力学平衡限制[5],反应方程式如下[方程(3)为主反应,方程(4)和方程(5)为副反应]:
(3)

(4)
(5)
催化剂的活性决定了选择氧化反应中硫磺的产率。V2O5、Fe2O3等金属氧化物广泛应用于H2S选择氧化反应中[6-7]。但是,反应过程中需要过量的氧气,且不耐氧气浓度的波动,从而制约了其在实际中的使用。二氧化铈具有优异的氧化还原能力和较高的储氧能力(OSC),广泛应用于环境催化领域[8-10]。近年来,铈基催化剂应用于硫化氢选择氧化也得到了越来越广泛的研究[11-13]。Zhang Fenglian等[11]研究发现,Ce基MgAlCeO水滑石衍生复合氧化物对于硫化氢选择氧化反应具有良好的催化活性,在140 ℃时可达到95%的硫回收率。Zheng Xiaohai等[13]研究了不同晶面二氧化铈催化H2S选择氧化活性的影响,结果表明,具有(100)和(110)晶面的CeO2纳米棒具有最优异的催化活性。
二氧化铈在高温下易烧结,热稳定性较差。通过在二氧化铈中掺入二氧化锆,可形成铈锆固溶体(CexZr1-xO2)[14-15]。Zr的掺入可以增强催化剂的热稳定性,显著提升其氧化还原能力和储氧能力(OSC)。铈锆固溶体是一种优异的催化材料[16-17],广泛应用于CO氧化、汽车尾气三效催化剂、水煤气变换等反应。然而,目前还没有将铈锆固溶体应用于H2S选择氧化的相关报道。
基于上述分析,本文将铈锆固溶体用于H2S选择氧化过程。一方面,利用Ce3+/Ce4+较低的氧化还原电位提升催化剂的低温活性;另一方面,铈锆固溶体较强的储放氧能力使得反应过程中不需要大量的氧气,更耐氧波动,Ce/Zr比例显著影响铈锆固溶体的催化活性。因此,考察Ce/Zr比例对催化剂结构和物化性质以及催化H2S选择氧化反应活性的影响,并且对反应机理进行研究。
1 实验部分
1.1 试 剂
硝酸铈、硝酸锆和氨水,均为分析纯,国药集团化学试剂有限公司;去离子水,自制。
1.2 催化剂制备
采用共沉淀法制备不同Ce/Zr物质的量比的CexZr1-xO2(x=0.9、0.7、0.5、0.3、0.1)复合氧化物。首先将Ce(NO3)3·6H2O和Zr(NO3)3·5H2O按物质的量比溶于50 mL去离子水中。向上述溶液中剂缓慢滴加质量分数3.6%的氨水,将溶液pH值调至9~10后,搅拌0.5 h,静置陈化12 h。将得到的沉淀抽滤后洗涤,滤饼于110 ℃在空气中干燥过夜,550 ℃煅烧4 h,制得铈锆固溶体CexZr1-xO2。
1.3 催化剂表征
比表面积和孔结构采用美国麦克仪器公司的ASAP-2020型吸附仪在液氮温度-196 ℃下测定。进行分析测定之前,催化剂在300 ℃下真空脱气预处理2 h。
XRD表征采用PANalytical X’Pert PRO型粉末衍射仪,Cu Kα(λ=0.15 418 nm),工作电流为40 mA,工作电压为40 kV,扫描范围为10°~80°。
拉曼(Raman)表征采用具有共聚焦显微镜的模块化多通道拉曼光谱系统(JY-HR800)。扫描范围为(50~800) cm-1,光谱分辨率为1 cm-1。所有的测量均在室温下进行。
X射线光电子能谱(XPS)表征采用Thermo ESCALAB 250型仪器,Al Kα,工作电压为5×10-8Pa,结合能使用内标碳1s峰(Eb=285 eV)进行校准,精度±0.2 eV。
H2程序升温还原(H2-TPR),O2程序升温脱附(O2-TPD)和CO2程序升温脱附(CO2-TPD)表征均采用美国麦克仪器公司Auto Chem 2720型仪器。在O2-TPD测定中,称取约0.1 g样品,在氦气流(50 mL·min-1)下400 ℃预处理1 h,冷却至室温后,通入稳定的O2/He(50 mL·min-1)直至吸附饱和。最后,以10 ℃·min-1从50 ℃至850 ℃进行O2脱附。CO2-TPD分析方法与前述相似,先进行预处理,吸附CO21 h后,在(50~850) ℃范围内进行CO2的脱附。在H2-TPR的测定中,称取约 0.1 g 样品,氦气气氛下(50 mL·min-1)在400 ℃预处理1 h,然后冷却至室温。程序升温还原过程为在H2/Ar(50 mL·min-1)气氛下,以10 ℃·min-1从50 ℃升至850 ℃。
1.4 催化剂活性评价
催化剂活性在常压下的固定床反应器中测定,尾气中H2S和SO2的浓度由装有FPD检测器的气相色谱在线分析。将0.5 mL的(20~40)目催化剂置于反应管中部,两端用石英棉进行封堵。反应气为0.05%(物质的量分数)H2S和0.025%(物质的量分数)O2,均用He平衡。体积空速为7 500 h-1,反应温度控制在(160~260) ℃。反应管后接冷凝管用于冷凝硫单质。
2 结果与讨论
2.1 CexZr1-xO2催化剂的物相和结构参数
图1为CexZr1-xO2催化剂的氮气吸附-脱附曲线和BJH方法计算的孔径分布曲线。表1为CexZr1-xO2催化剂的孔结构参数。由图1可知,铈锆复合氧化物均为典型的Ⅳ型等温线,当x>0.5时,具有H1型回滞环,为圆柱形的介孔材料;随着锆掺杂量的增加,回滞环逐渐变为H2型,意味着其具有“墨水瓶”型孔结构[18]。由表1可知,催化剂平均孔径(3.44~10.8) nm,并且随着Ce/Zr比例增加,孔体积与平均孔径整体上呈现增加的趋势。这可能是由于在铈锆固溶体形成过程中,Ce4+原子尺寸较大,更易导致晶格膨胀和孔结构的改变[19]。较大的孔径与孔体积可能有利于H2S在催化剂表面的扩散和传质,从而提高催化活性。然而,比表面积随着Ce/Zr比例增加略有降低,这可能是由于Zr离子的掺入增强了催化剂的热稳定性,使其更容易保持较高的比表面积。

图1 催化剂CexZr1-xO2的氮气吸脱-附等温线和BJH孔径分布图Figure 1 Nitrogen adsorption-desorption isotherms and pore size distribution of the CexZr1-xO2 catalysts

表1 催化剂CexZr1-xO2的结构参数Table 1 Texturalproperties of CexZr1-xO2catalysts
图2为CexZr1-xO2(x=0.9、0.7、0.5、0.3、0.1)的XRD图。由图2可知,CexZr1-xO2衍射峰的2θ值为29.8°、34.3°、49.6°和58.9°,分别对应(111)、(200)、(220)、(311)、(222)晶面,与立方相二氧化铈(PDF 28-1436)吻合。CexZr1-xO2催化剂具有立方结构的晶相,表明铈离子和锆离子均匀分布在晶体结构中,形成了铈锆固溶体[20]。通过谢乐公式计算的晶粒尺寸见表1,由表1可知,ZrO2的掺入使CeO2发生相位扭曲,半峰宽增加,晶粒尺寸减小。随着Ce/Zr比例的增加,2θ值向低角度移动,这是由于Zr4+(0.084 nm)离子半径小于Ce4+(0.097 nm)的离子半径,Zr4+掺入晶格后晶面间距缩小,表明铈锆固溶体的形成[21]。

图2 催化剂CexZr1-xO2的XRD图Figure 2 XRD patterns of CexZr1-xO2 catalysts
铈锆固溶体中可能存在3种稳定相[单斜(m)、四方(t)或立方(c)]和2种亚稳相(t′、t″),仅通过XRD无法获得确切的晶体结构。因此,采用拉曼光谱进一步确定物相的精细结构,结果如图3所示。由图3可知,461 cm-1和600 cm-1的峰分别归属于Ce-O键的对称伸缩F2g拉曼活性模式和氧空位[22]。随着x值的增加,F2g峰逐渐发生红移,且半峰宽变大,这是因为Zr4+掺入CeO2晶格内,导致晶格畸变的发生,使晶粒尺寸发生变化[23-24],进一步确定了铈锆固溶体的生成。当x=0.7和0.9时,137 cm-1、245 cm-1、309 cm-1和625 cm-1附近出现4个弱峰,来自立方氧化锆的拉曼活性模式(A1g + 2B1g + 3Eg)[25],意味着t″亚稳相结构的存在[26]。值得注意的是,461 cm-1附近的特征峰依然存在,表明固溶体的立方萤石结构依然保持。

图3 催化剂CexZr1-xO2的Raman谱图Figure 3 Raman spectrum of CexZr1-xO2 catalysts
2.2 CexZr1-xO2催化剂的表面化学态与表面氧
采用XPS对催化剂表面的化学态进行分析,结果如图4所示。图中V与U分别对应Ce 3d5/2和Ce 3d3/2的自旋分裂轨道[27]。由图4可知,Ce 3d轨道可以被分峰为8个峰:U‴(~917.5 eV),U″(~907.5 eV),U′(~903.5 eV),U(~901.4 eV),V‴(~898.9 eV),V″(~888.9 eV),V′(~884.6 eV)和V(~882.5 eV)。表明Ce4+和Ce3+同时存在于催化剂表面,其中,Ce4+标记为U‴、U″、U、V‴、V″和V,Ce3+标记为U′和V′。通过峰面积进行半定量计算,可以得出Ce3+占Ce3++Ce4+的物质的量比例,结果列于表2。由表2可知,Ce0.9Zr0.1O2、Ce0.7Zr0.3O2、Ce0.5Zr0.5O2、Ce0.3Zr0.7O2和Ce0.1Zr0.9O2中Ce3+占Cetotal的物质的量比分别为16.8%、22.1%、21.0%、22.4% 和24.3%。据文献报道,Ce3+的存在可以促进表面氧空位的生成,因此,氧空位的浓度随Ce3+的增多而增大[28]。由图4还可知,O 1s轨道可分为在530.1eV附近和532.5 eV附近的两个峰,分别对应晶格氧(Olat)和氧空位上的化学吸附氧(Oads)。根据峰面积进行定量计算得到,Ce0.9Zr0.1O2、Ce0.7Zr0.3O2、Ce0.5Zr0.5O2、Ce0.3Zr0.7O2和Ce0.1Zr0.9O2化学吸附氧的物质的量比分别为30.0%、30.1%、35.4%、27.4%和36.8%,这与Ce 3d XPS结果基本吻合。

图4 催化剂CexZr1-xO2的Ce 3d 和O 1s谱图Figure 4 Ce 3d and O 1s XPS spectra of the CexZr1-xO2 catalysts

表2 催化剂CexZr1-xO2的XPS结果分析Table 2 XPS analysis of the CexZr1-xO2 catalysts
图5为催化剂CexZr1-xO2的O2-TPD谱图。
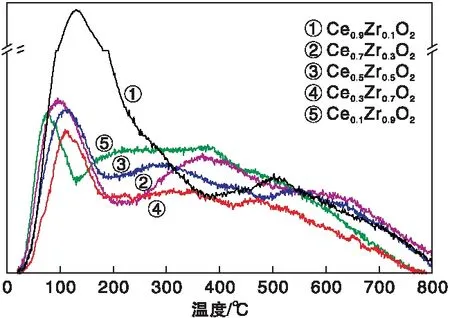
图5 催化剂CexZr1-xO2的O2-TPD谱图Figure 5 O2-TPD spectra of the CexZr1-xO2 catalysts
由图5可知,铈锆固溶体的表面上存在3种O2脱附峰,200 ℃以下的脱附峰归属于氧空位上物理吸附氧的脱附,(200~500) ℃为化学吸附的表面氧物种的脱附,而500 ℃以上则对应晶格氧的脱附[29]。对于物理吸附氧,随着Ce/Zr比例的增加,脱附峰面积增大,脱附温度升高,意味着催化剂对氧气的吸附增强,这可能是与氧空位的浓度或掺杂导致的结构改变有关[30]。但化学吸附氧与晶格氧的脱附峰差别不大,Ce0.1Zr0.9O2的化学吸附氧含量相对较高,而Ce0.9Zr0.1O2和Ce0.7Zr0.3O2的化学吸附氧含量则相对较低,Zr的掺杂有利于氧空位的增加,结果与XPS分析相一致。
2.3 CexZr1-xO2催化剂的表面碱性
对于硫化氢选择氧化反应,硫化氢在催化剂表面碱性位的吸附对催化活性至关重要。因此,通过CO2-TPD来评价CexZr1-xO2催化剂表面的碱性,结果如图6所示。
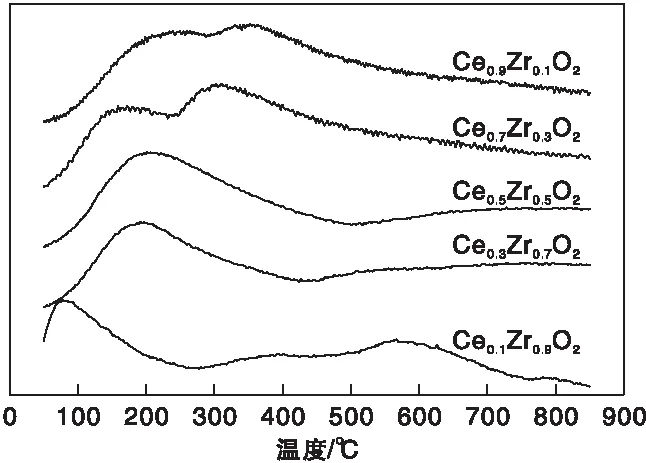
图6 催化剂CexZr1-xO2的CO2-TPD谱图Figure 6 CO2-TPD spectra of the CexZr1-xO2 catalysts
催化剂表面的碱性位可根据脱附温度分为3类:弱碱性位[(50~200) ℃]、中度碱性位[(200~400) ℃]和强碱性位 [(400~600) ℃][24]。由图6可知,以峰面积作为半定量的依据,随着Zr的掺入,弱碱性位不断增多,而中度碱性位减少,且脱附峰逐渐向低温方向移动,碱性强度降低。因此,Ce0.9Zr0.1O2具有更多的中度碱性位且碱性较强。根据文献报道[13],在硫化氢选择氧化过程中,硫化氢更倾向于吸附在中度碱性位上,再发生反应,因此,中度碱性位的存在对硫化氢选择氧化反应至关重要。
2.4 CexZr1-xO2催化剂的氧化还原性
图7为催化剂CexZr1-xO2的TPR曲线。在铈锆固溶体中,H2的消耗均归因于Ce4+到Ce3+的还原,而Zr4+不被还原[31]。由图7可知,对于Ce0.9Zr0.102、Ce0.7Zr0.3O2和Ce0.5Zr0.5O2,其峰型与CeO2类似,在500 ℃和800 ℃处有2个峰,分别对应着表面和体相中Ce4+的还原[32]。随着Zr的增加,表面还原与体相还原的峰面积之比不断增加,对于Ce0.3Zr0.702和Ce0.1Zr0.9O2只有在(500~600) ℃之间一个消耗峰,表明Zr的掺入有利于提高晶格中氧的移动速率,使晶格氧可以快速迁移到催化剂表面,此时,还原不仅局限于表面的Ce4+,体相的Ce4+也参与其中[33]。与此同时,随着Zr含量的增加,低温区的消耗峰向高温移动。推测这是由于Zr的含量增加使得晶体中不可还原的组分增加导致,此外,当x<0.5时,由于Ce和Zr的相互作用使晶体结构发生改变,铈锆固溶体的结构以四方晶系为主,更难以被还原。因此,Ce0.9Zr0.1O2具有最强的氧化还原能力。

图7 催化剂CexZr1-xO2的H2-TPR曲线Figure 7 H2-TPR curves of the CexZr1-xO2 catalysts
2.5 反应温度对CexZr1-xO2催化活性的影响
反应温度对不同Ce/Zr比例的铈锆固溶体催化硫化氢选择氧化反应转化率、选择性和产率的影响如图8所示。由图8可以看出,在反应温度(160~260) ℃内,随着温度增加,所有催化剂的转化率均先增高后趋于稳定。其中,Ce0.9Zr0.1O2有出色的低温活性,在(160~260) ℃全温度区间内,转化率均保持在95%以上。并且,在反应温度180 ℃时就可达到最高转化率98%,Ce0.7Zr0.3O2、Ce0.5Zr0.5O2和Ce0.1Zr0.9O2则在反应温度220 ℃达到最高转化率,Ce0.3Zr0.7O2在反应温度240 ℃才能达到最高转化率。Ce0.9Zr0.1O2在所有温度范围内均表现出较稳定且高于95%的转化率。

图8 反应温度对CexZr1-xO2催化剂的转化率、选择性和产率的影响Figure 8 Effect of temperature on H2S conversion,selectivity and sulfur yield for CexZr1-xO2 catalysts
由图8还可以看出,随着反应温度的升高,硫选择性略有降低。反应温度为180 ℃时,Ce0.9Zr0.1O2和Ce0.7Zr0.3O2的选择性开始下降,分别为99%和97%。当反应温度升至220 ℃以上时,各催化剂的选择性几乎相当。但在整个温度区间内,所有催化剂的选择性均保持在95%以上,选择性差距不大,催化剂的硫收率与其转化率变化趋势一致。因此,铈锆固溶体催化硫化氢选择氧化的催化活性基本随着Ce/Zr比的增加而提高,Ce0.9Zr0.1O2具有最高的催化活性,在180 ℃时收率可达到97%。
结合催化剂的结构和性质分析,虽然Zr的掺入使催化剂的表面积增加,但孔径变小,且孔结构变为墨水瓶形,而Ce0.9Zr0.1O2具有较大的孔体积和孔径,同时拥有最多的中度碱性位,有利于反应气体在催化剂表面的传质与吸附。所有的催化剂均具有充足的氧空位和晶格氧迁移速率,因此,均可以在n(H2S):n(O2)=2∶1时达到95%以上的收率。Ce4+为催化反应的活性中心,Ce0.9Zr0.1O2最高的低温活性主要归因于其最高的Ce4+含量与最强的氧化还原能力。
2.6 催化反应机理
为了探究铈锆固溶体催化H2S选择氧化反应的机理,对反应后的催化剂进行XPS分析,考察催化剂表面元素的变化情况,结果如图9所示,通过峰面积半定量计算得到的Ce3+和Oad含量见表2。由图9和表2可知,反应后,Ce3+的含量从16.8%增加至18.3%,表明Ce4+作为活性组分参与到反应中。同时,反应后的晶格氧被消耗,表明晶格氧参与催化反应过程中,并在反应过程中被消耗。

图9 催化剂CexZr1-xO2反应后的Ce 3d、O 1s和S 2p谱图Figure 9 Ce 3d,O 1s and S 2p XPS spectra of the used CexZr1-xO2 catalysts
基于上述结果,推断反应机理如下:(1)H2S吸附在催化剂表面的中碱性位上;(2)Ce4+将吸附的H2S氧化成单质硫,同时自身被还原为Ce3+;(3)晶格氧迁移至催化剂表面,作为活性氧物种将Ce3+氧化为Ce4+;(4)活性氧物种通过吸附气态氧得到补充。

3 结 论
(1) 通过共沉淀法制备了一系列含有不同Ce/Zr物质的量比的铈锆固溶体,考察了Ce/Zr物质的量比对晶体结构、碱性和氧化还原性的影响,并测试了其在硫化氢选择氧化反应中的催化活性。
(2) 所有催化剂在n(H2S)∶n(O2)=2∶1时均可以在一定温度下达到95%以上的硫磺收率。催化剂的低温催化活性随着Ce/Zr比例的减少呈下降趋势,其中Ce0.9Zr0.1O2具有最高的催化活性,在(160~260) ℃均保持较高的转化率,同时在较低的温度下(180 ℃))即可达到97%的最佳收率。结合表征推测,一方面是因为Ce0.9Zr0.1O2催化剂具有较大的孔径与孔体积,较强的中度碱性位,有利于传质和吸附,另一方面,其Ce4+的含量最高,具有最强的氧化还原能力。
(3) 推测了催化反应的机理以及失活原因,反应遵循氧化还原机理,催化剂的失活主要是因为表面生成硫酸盐物种消耗了活性组分。
