先天性无阴道综合征超声诊断研究*
2020-11-27芦爱霞
芦爱霞,李 群,李 梅
1.深圳市人民医院超声科,广东 深圳 518000;2.深圳市罗湖区人民医院超声科,广东 深圳 518000
先天性无阴道综合征(Mayer-Rokitansky-Küster-Hauser Syndrome MRKH 综合征)发生率较低,为活产女婴的1/4500[1],本文回顾性分析了深圳市罗湖区人民医院2009年8 月—2018 年5 月共501 例MRKH 综合征患者均已行经腹腔镜腹膜成形术(罗湖术式)的病例,与手术结果对照,总结其超声表现,以期提高超声医生对本病的认识和诊断水平。
1 资料与方法
1.1 一般资料
收集深圳市罗湖区人民医院2009 年8 月—2018 年5 月MRKH综合征病例501例,均已行经腹腔镜腹膜成形术(罗湖术式),患者年龄18~45 岁,平均年龄(23.6±6.8)岁,初诊年龄13~20岁,回顾性分析超声检查患者子宫附件区及泌尿系的检查结果。超声诊断仪型号为GE E8、GE E6、Philips IU22及日立5500等。
1.2 统计学方法
本组资料采用SPSS 17.0 软件进行统计分析,超声检查发现始基子宫和卵巢数量与手术结果比较采用卡方检验,以P<0.05表示差异有统计学意义。
2 结果
2.1 生殖系统畸形及病变
501 例MRKH 综合征患者行超声检查均未见阴道回声;192 例未见始基子宫,309 例可见始基子宫(见图1)。而术中证实有一例左侧未见始基子宫,合并同侧肾脏异位于盆腔;一例左侧未见始基子宫,合并同侧多发结石并积液;一例双侧未见始基子宫,仅见盆底膀胱直肠间条索状纤维带痕迹,合并右肾缺如;其余病例均可见双侧始基子宫,位于附件旁,大小为1.2~4.5 cm,中间以索状纤维带连接。
本组资料超声显示451 例可见双侧卵巢(见图2),25例可见单侧卵巢,25例双侧卵巢显示均欠满意。术中证实先天性无阴道综合征患者合并卵巢及输卵管发育异常9例,其中有三例单侧肾缺如病例合并同侧输卵管和或卵巢缺如,仅见痕迹;一例右肾缺如合并左肾双肾盂,可见卵巢未见输卵管;两例有单侧附件发育不良,卵巢均为1.0 cm×1.0 cm,输卵管缺如或仅见伞端;一例左卵巢正常,右卵巢及双输卵管缺如;一例有输卵管仅见伞端;一例右侧输卵管卵巢缺如,可见始基子宫,合并左侧1-2 肋融合并胸椎发育畸形。
2.2 泌尿系统病变
501 例MRKH 综合征发育异常患者有460 例可查到肾脏超声检查结果,发现肾脏发育异常39 例。其中肾缺如19 例;肾脏异位至盆腔8 例,其中一例肾脏异位合并对侧肾脏双肾盂;马蹄肾3 例;另有一例病人因单侧肾脏畸形已行肾脏切除;2 例单侧肾多发肾结石,1 例双肾多发结石;5例肾囊肿。

图1 超声显示可见始基子宫
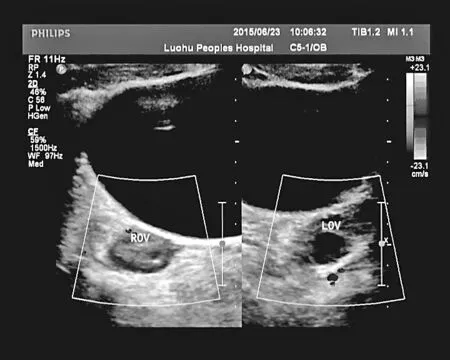
图2 超声显示可见双侧卵巢
2.3 染色体异常
501例患者142例可见染色体检查结果,有7例发现异常,其中一例为Turner 综合征,染色体为45X/46XX;一例为45X;一例为46XXY;2 例染色体为46XX qqh+;两例为46XX G带。其余染色体均为46XX。
2.4 临床表现
乳房及第二性征开始发育时间为9~18 岁,平均13.5岁。均表现为青春期无月经来潮,6 例有周期性下腹痛,其余病例均未见明显不适。
3 讨论
MRKH 综合征的胚胎发育基础是副中肾管不发育或发育不良,阴道不能形成,子宫发育不良,可有正常的输卵管和卵巢。因青春期原发性闭经被临床诊断,其病因不明,大多成散发发病,是包括子宫及阴道发育不全及肾脏、骨骼、听力缺陷等多发畸形的综合病征。 本组资料显示,MRKH 综合征手术年龄为(23.6±6.8)岁,与文献报道基本一致[2-3]。超声检查192例未能显示始基子宫,而术中证实仅有3 例合并单侧或双侧始基子宫缺如,其余均可见双侧始基子宫;而导致超声漏诊的原因为部分始基子宫较小、始基子宫位置较高或位于髂窝处,受盆腔肠气的影响而显示不清晰;或者MRKH 综合征在临床较少见,超声医生不了解MRKH 综合征的超声表现,影响了对始基子宫的观察。
术中发现有4 例始基子宫合并子宫肌瘤,3 例合并子宫腺肌症,12 例合并卵巢囊肿,3 例合并畸胎瘤,3 例合并巧克力囊肿,8 例合并系膜囊肿,双骶韧带表面腹膜囊肿一例,右卵巢赘生物0.5 cm(病理为卵泡膜细胞呈瘤样增生)[4-6]。
本组资料超声显示90%(451/501)可见双侧卵巢,4.9%(25/501)可见单侧卵巢,4.9%(25/501)双侧卵巢显示均欠满意, 部分卵巢未能显示考虑主要原因是卵巢异位后位置改变及肠气较多。术中证实1.8%(9/501)MRKH 综合征患者合并卵巢及输卵管发育异常,有两例患者合并腹股沟疝,疝内容物为同侧的卵巢、输卵管,对于MRKH 综合征患者如合并腹股沟区肿物,应考虑到疝内容物为同侧附件的可能性[3,7]。合并肾脏畸形发生率为8.5%(39/460),其中肾缺如4.1%(19/460);肾脏异位至盆腔1.7%(8/460),马蹄肾0.6%(3/460);0.6%(3/460)肾多发肾结石;1.0%(5/460)肾囊肿。MRKH综合征检查肾脏的意义主要是对于单侧肾缺如者应注意保护健侧肾功能,对于异位的肾脏避免因为盆腔包块而切除。发现肾脏畸形对于确诊先天性无阴道综合征有一定的意义,因二者在胚胎发育过程中密切联系[8]。有6 例患者合并周期性下腹痛,超声检查发现一侧或双侧始基子宫较大,可见内膜;其中两例患者盆腔可见混合性包块,周边可见肌层包绕,未见宫颈及阴道,均合并肾脏发育畸形,术中证实为功能性始基子宫,合并盆腔子宫内膜广泛异位、粘连。对于无月经来潮,有周期性下腹痛表现患者应及时进行超声检查,需与阴道闭锁及处女膜闭锁等疾病鉴别。
142 例可见染色体检查结果中染色体异常率16.7%(7/142),其余染色体均为46XX。2011 年有研究显示[9],MRKH 综合征可能与胚胎发育相关候选基因、转录、甲基化过程及孕期母体内分泌异常等综合因素相关。本组资料统计存在的缺陷是患者染色体检查数量尚不多。
MRKH 综合征主要需与阴道闭锁相鉴别,阴道闭锁分为四型I 型表现为阴道下段或中段闭锁,其上阴道及子宫发育正常,II 型变现为阴道完全闭锁,合并宫颈闭锁,子宫内膜可有正常分泌功能,III型闭锁表现为阴道上段或中上段闭锁,合并宫颈闭锁,子宫内膜可有正常分泌功能;IV型闭锁表现为阴道顶端闭锁,合并宫颈闭锁者,子宫内膜可有正常分泌功能。阴道闭锁I型、III型及IV 型因有部分阴道可见,易与MRKH 综合征相鉴别。而II型阴道闭锁与MRKH 综合征鉴别较困难,二者鉴别点在于前者是尿生殖窦发育缺陷所致,合并宫颈发育异常,子宫体为单个,发育正常或虽有畸形但有功能性内膜[10],而后者无阴道,有双侧始基子宫,子宫绝大多数无功能性内膜,对于有功能性内膜的先天性无阴道综合征及II型阴道闭锁的患者确诊后应尽快手术治疗以缓解症状,对于无功能性内膜的先天性无阴道综合症患者应18岁成年后[9]择期手术。
综上所述,本组资料为超声诊断MRKH 综合征提供了子宫、卵巢及泌尿系统常见畸形的超声表现及其发生率,为超声医生诊断本病提供了有意义的参考价值,以期为临床医生提供更加全面、准确的超声检查结果。
