金红石型TiO2(110)表面与Ag相互作用的第一性原理研究
2020-11-24张丽仙许彦亭郭俊梅
鲍 冰,沈 月,张丽仙,潘 勇,许彦亭,郭俊梅,闻 明 *
金红石型TiO2(110)表面与Ag相互作用的第一性原理研究
鲍 冰1,沈 月2,张丽仙3,潘 勇4,许彦亭2,郭俊梅2,闻 明2 *
(1. 贵研金属(上海)有限公司,上海 200050;2. 昆明贵金属研究所 稀贵金属综合利用国家重点实验室,昆明 650106;3.云南省第一人民医院,昆明理工大学附属医院,昆明 650032;4. 西南石油大学,成都 610002)
采用第一性原理计算Ag与金红石型TiO2(110)晶面的相互作用。通过计算金红石型TiO2(110)面上2种键桥对Ag原子的吸附能,发现键桥Ti形式与Ag原子结合的吸附能低于键桥O形式的吸附能;通过进一步的电荷布居、单位键长以及电子结构的计算,发现键桥Ti形式的Ti-Ag之间的结合能大于键桥O形式的Ti-Ag之间的结合能,这说明Ag原子更容易与金红石型TiO2(110)面上的键桥Ti上的Ti原子发生反应生成相应的化合物。
计算材料学;第一性原理;Ag;金红石型TiO2;键桥;电子结构
钛材料因其低密度、高比强度、优异的抗腐蚀性、无磁性、加工性好等特点,广泛用于航空航天、航海、医疗、交通等领域[1-2]。但是,钛材料耐磨性差、易擦伤及粘附限制了其推广应用。材料的磨损、疲劳和腐蚀等失效大多开始于表面,表面结构状态对材料的使用寿命具有重要的影响。为了延长材料使用寿命和满足特殊环境的使用要求,通过提高材料表面性能从而提高材料整体性能成为关键。对钛材料进行表面改性以增强其耐磨性,同时保持良好的耐腐蚀性成为该领域一个研究热点[3]。Au、Ag在碳化物、氮化物、氧化物、金属合金中均具有良好的润滑性能[4]。Lince等[5]采用共溅射技术制备出纳米Au粒子/MoS2涂层,涂层在低接触压力下具有低电阻、低摩擦系数等特点适合作为集电环、开关元件。Cui等[6]发现添加了一定量Ag的青铜在摩擦实验后,磨损表面形成了一层Ag润滑膜,明显降低了青铜的摩擦系数,从而提高了其在海水环境下的抗磨损性能。Hou等[7]研究结果表明,添加微量的Ag可有效提高Ti的抗腐蚀性。因此,设想可以在钛材料的表面制备出一层Ag/TiO2复合层,通过使具有较高耐磨性的TiO2和具有良好塑性的Ag相结合,获得高强高润滑的耐磨表面,从而提高钛材料的耐磨性。
常见的TiO2有板钛矿、锐钛矿型和金红石型,曹红红等[8]计算了掺Sn的锐钛矿TiO2的电子结构,结果表明,掺杂后Sn-O键比原来纯锐钛矿的Ti-O键强。金红石作为TiO2的稳定相受到了人们的广泛关注,但是Ag与金红石型TiO2的第一原理计算相对很少。针对Ag/金红石型TiO2复合涂层是否会发生相互作用,本文采用第一性原理进行计算,以研究其微观相互作用机理。
1 计算方法
考虑到计算周期和目前实验中遇到的最常见的金红石型TiO2晶面,本文采用金红石型TiO2的最强峰(110)面进行了计算。金红石型TiO2(110)面上存在2种键桥形式:Ti键桥和O键桥。考虑到这2种键桥吸附Ag原子的理论计算,本文采用基于第一性原理的密度泛函理论(DFT)平面波赝势方法,所有的计算都是在CASTEP模块中完成的。在Kohn-Sham能量泛函形式中,电子之间的交换关联能是以电子密度的形式表达的。交换关联势选取的是局域密度泛函势(LDA),通过CA-PZ函数进行交换相关势的修正,对金红石型TiO2(110)面吸附Ag原子的结构模型进行几何优化,动能截至能取为360 eV,K点取6×6×4,结构优化采用的是基于Pulay的密度混合方案,SCF误差为2.0×10-6eV,其它参数一律选中等精度。通过计算晶格常数、结合能、电子布居、单位键长以及电子结构等参数,从理论上研究了Ag/TiO2复合材料中的Ag与TiO2(110)面的相互作用。
2 结果与讨论
2.1 两种键桥上Ag原子的基本参数
2.1.1吸附能
由于金红石型TiO2(110)面上存在Ti键桥和O键桥2种键桥形式,自然Ag在不同的键桥形式下其吸附能也是不一样的,吸附能的差异必然会导致Ag在不同的吸附位置其稳定性不同。首先通过吸附能来判断金红石型TiO2(110)面上吸附Ag原子的稳定性,吸附能的计算公式为:
ad=total-(TiO2(110)+atom Ag) (1)
其中,total为金红石型TiO2(110)面上吸附Ag原子的总晶胞能,TiO2(110)为金红石型TiO2(110)面的晶胞能,而atom Ag为单个Ag原子的基态能。
表1为金红石型TiO2(110)面上2种键桥形式的吸附能。从表1中可以看得出,键桥O吸附Ag原子的吸附能为-2.78 eV,键桥Ti位置吸附Ag原子的吸附能为-2.81 eV。由此可见,键桥Ti吸附Ag原子的吸附能要小于键桥O的吸附能,也就是说当金红石型TiO2(110)面上吸附Ag原子时,更容易以键桥Ti的方式与Ag原子结合。
表1 Ag在金红石型TiO2(110)不同键桥方式的吸附能

Tab.1 The adsorption energy of Ag on rutile (110) with different bridge types
2.1.2电子占据数、电荷布居和单位键长
表2~表4分别为金红石型TiO2(110)、键桥O吸附Ag原子、键桥Ti吸附Ag原子模型各原子轨道上的电子占据数。
表2 纯金红石型TiO2(110)上各原子轨道上的电子占据数

Tab.2 Electron occupancy in each atomic orbital of pure rutile(110)
表3 键桥O上各原子轨道上的电子占据数

Tab.3 Electron occupancy in each atomic orbital of O bridge
表4 键桥Ti上各原子轨道上的电子占据数
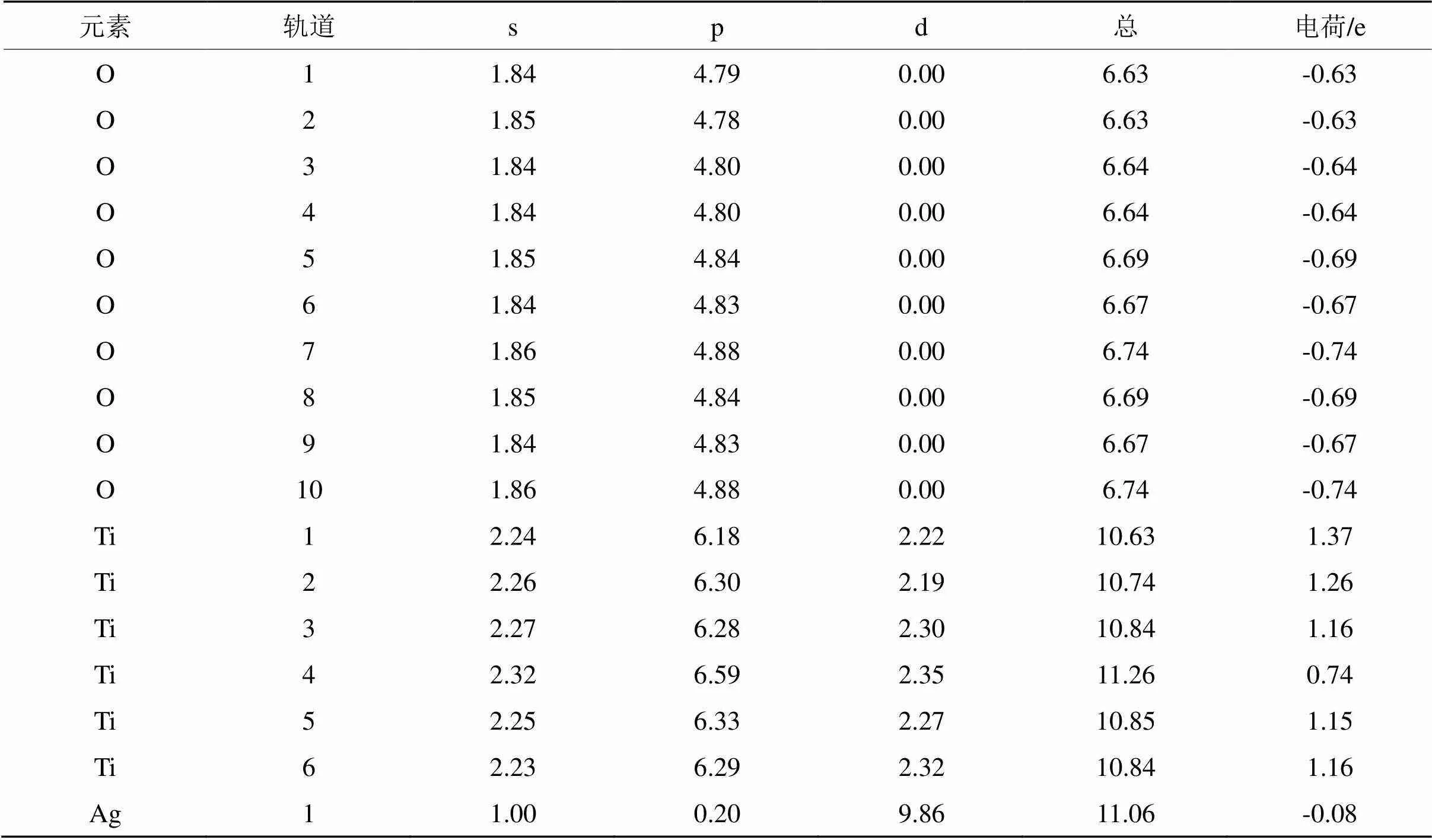
Tab.4 Electron occupancy in each atomic orbital of Ti bridge
从表2~表4可以看出,虽然各原子轨道电荷布居发生了变化,主要是O原子得到电子,而Ti原子失去电子,当金红石型TiO2(110)面吸附Ag原子时,Ag原子也是得到电子。从整体而言,外层轨道电荷总数基本是保持不变,这遵守了电子守恒准则,但可以看出各原子外层轨道上得失电子数是不一样的,表明外层轨道上电子发生了spd杂化。值得注意的是,无论是键桥O形式还是键桥Ti形式的吸附,在吸附过程中Ag原子均是得到了电子。这也意味着当金红石型TiO2(110)吸附Ag原子时,将是Ti-Ag之间的电子转换。
表5~表7为金红石型TiO2(110)、键桥O吸附Ag原子以及键桥Ti吸附Ag原子模型的电荷布居和单位键长。
表5 纯金红石型TiO2(110)的电荷布居和单位键长
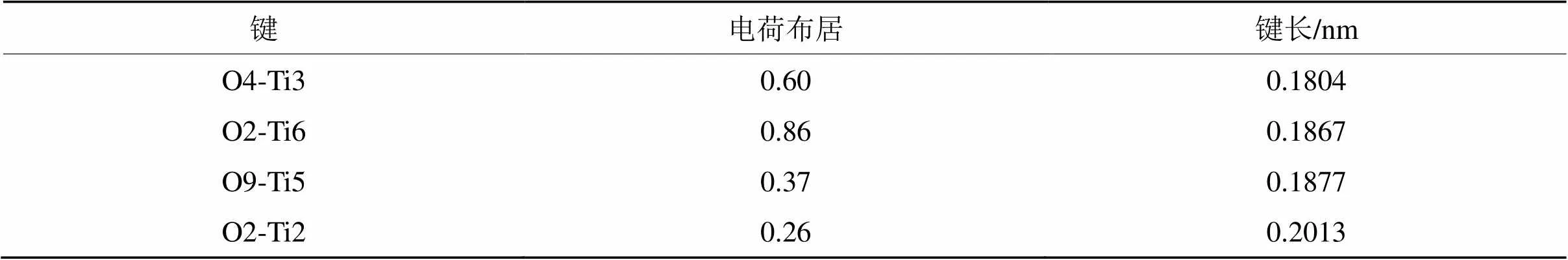
Tab.5 Bond lengths and population of pure rutile (110)
表6 键桥O形式的电荷布居和单位键长
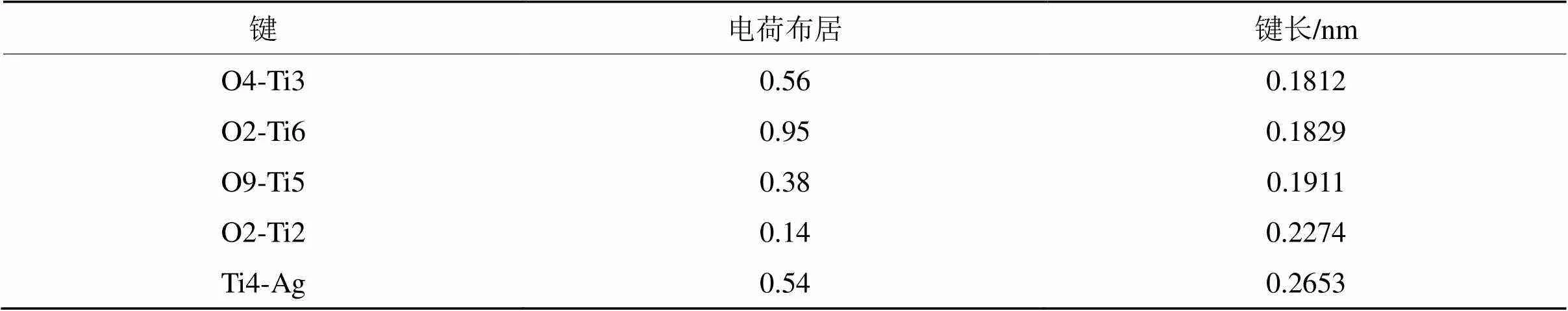
Tab.6 Bond lengths and population of O bridge

表7 键桥Ti形式的电荷布居和单位键长
从表5~表7可以看出,纯TiO2(110)面的Ti-O之间的电荷布居在0.86~0.26之间,而Ti-O之间的单位键长则为0.1804~0.2013 nm之间。当吸附Ag原子时,键桥O原子的Ti-Ag之间的电荷布居为0.54,单位键长为0.2653 nm,而键桥Ti原子的Ti-Ag之间的电荷布居为0.54,单位键长为0.2638 nm。2种键桥形式的电荷布居一致,但是键桥Ti原子的Ti-Ag之间的单位键长则略小于键桥O原子的单位键长,从理论上而言,键长越短则原子之间的结合能力就越强,从上面2种键桥形式的Ti-Ag之间的单位键长来看,键桥Ti更容易吸附Ag原子,这与键桥Ti吸附Ag原子的分析是一致。
2.2 两种键桥上吸附Ag原子的电子结构计算
为深入理解金红石型TiO2(110)面上吸附Ag原子的吸附性质,进一步从电子结构层次上分析它的吸附形式。原子之间相互作用的电子转移主要发生在各原子的价电子上,这几种元素的价电子分别为O 2s22p2、Ti 3p63d24s2和Ag 4p64d105s1。图1分别为金红石型TiO2(110)、键桥Ti形式吸附Ag和键桥O形式吸附Ag的总态密度分布图。图2为金红石型TiO2(110)各原子的分波态密度图,图3分别为键桥Ti形式吸附Ag和键桥O形式吸附Ag的各原子的分波态密度图,图中横坐标零点处为Fermi能级。
从图1~图3中可以看出,金红石型TiO2(110)的态密度分布图主要是由O的2p电子轨道上的电子和Ti的3d电子轨道上的电子贡献,当金红石型TiO2(110)吸附Ag原子时,则有Ag原子的4d电子轨道上的电子和部分的5s轨道上的电子参与贡献。对比图2和图3,发现在费米面附近键桥O形式的Ag原子的主峰峰值为8.50 eV,而键桥Ti形式的Ag原子的主峰峰值为8.40 eV,这说明键桥Ti形式的Ag原子失去的电子将多于键桥O性质的Ag原子电子,这说明键桥Ti形式的Ti-Ag之间将反应更为剧烈,即当金红石型TiO2(110)面吸附Ag原子时,Ag原子更容易与金红石型TiO2(110)面上Ti原子发生反应。
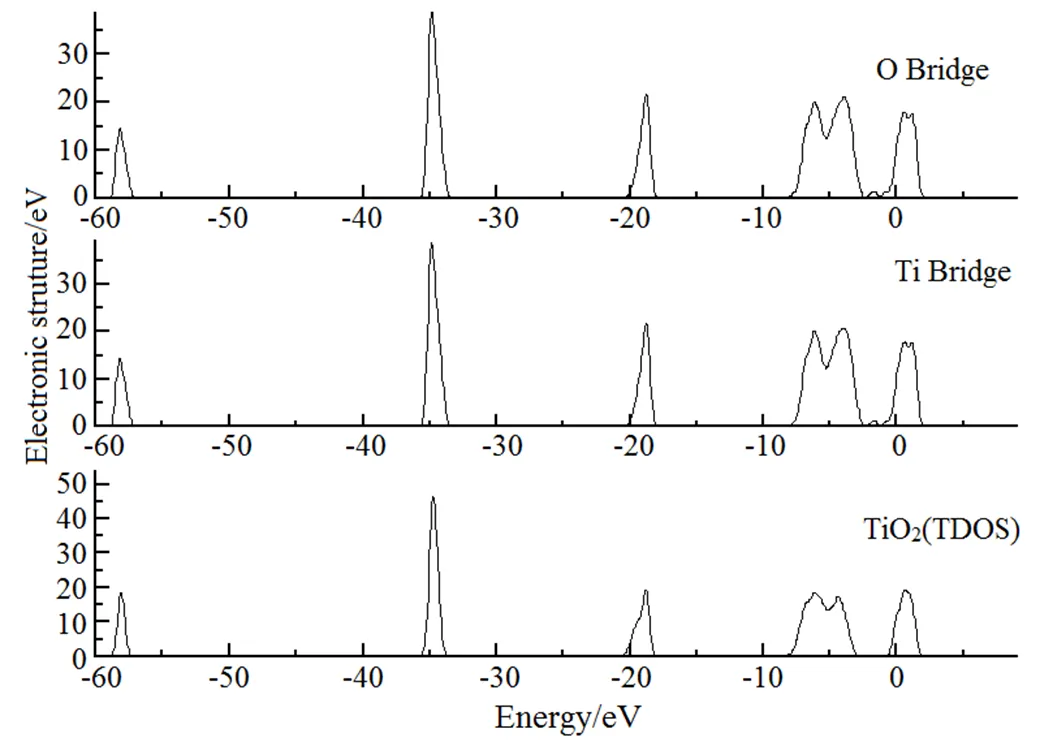
图1 金红石型TiO2(110)、键桥O和键桥Ti模型吸附Ag的总态密度分布图

图2 金红石型TiO2(110)中各原子的分波态密度图
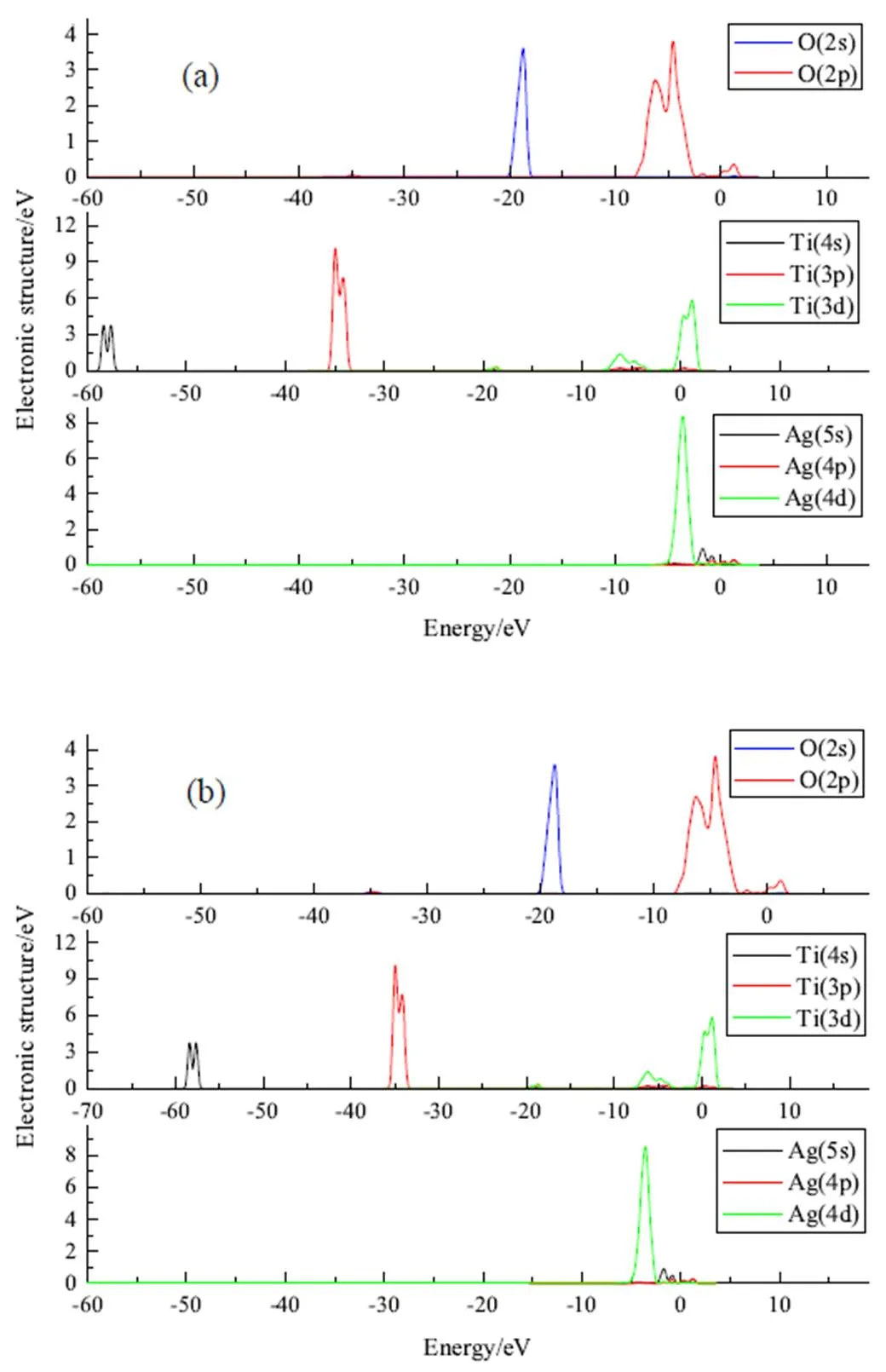
(a)O键桥(O bridge); (b). Ti键桥(Ti bridge)
3 结论
1) 对于金红石型TiO2(110)面,键桥Ti形式与Ag原子结合的吸附能低于键桥O形式的吸附能。
2) 电荷布居、单位键长以及电子结构的计算结果表明键桥Ti形式的Ti-Ag之间的结合能力高于键桥O形式的Ti-Ag之间的结合能力。
3) Ag原子更容易与金红石型TiO2(110)面上的键桥Ti上的Ti原子发生反应。
[1] 张蕊, 王鲁宁, 赵飞, 等. 贵金属在船用钛及钛合金表面改性中的应用[J]. 贵金属, 2017, 38(4): 74-80.
ZHANG R, WANG L N, ZHAO F, et al. Application of precious metals in the surface modification of marine titanium and titanium alloys [J]. Precious metals, 2017, 38(4): 74-80.
[2] 王鲁宁, 闻明, 张蕊, 等.含金耐磨耐蚀涂层的制备及性能研究进展[J]. 贵金属, 2019, 40(1): 75-81.
WANG L N, WEN M, ZHANG R, et al. Preparation and properties research progress of wear and corrosion resistance coatings containing gold[J]. Precious metals, 2019, 40(1): 75-81.
[3] WEN M, WEN C, HODGSON P, et al. Thermal oxidation behaviour of bulk titanium with nanocrystalline surface layer[J]. Corrosion science, 2012, 59: 352-359.
[4] MULLIGAN C P, BLANCHET T A, GALL D. CrN-Ag nanocomposite coatings: High-temperature tribological response[J]. Wear, 2010, 269(1/2):125-131.
[5] LINCE J R, KIM H I, ADAMS P M, et al. Nanostructural, electrical, and tribological properties of composite Au-MoS2coatings[J]. Thin solid films, 2009, 517(18): 5516-5522.
[6] CUI G J, BI Q L, YANG J, et al. The bronze-silver self-lubricating composite under sea water condition[J]. Tribology international, 2013, 60: 83-92.
[7] HOU L G, LI L, ZHENG Y F. Fabrication and characterization of porous sintered Ti-Ag compacts for biomedical application purpose[J]. Journal of materials science and technology, 2013, 29(4): 330-338.
[8] 曹红红, 陈强, 王天民. 掺Sn锐钛矿相TiO2的第一原理计算[J]. 稀有金属材料与工程, 2008, 37(2): 219-222.
CAO H H, CHEN Q, WANG T M. First principle calculations of the Sn doped anatase TiO2[J]. Rare metal materials and engineering, 2008, 37(2): 219-222.
A First Principle Study of the Interaction of Ag and Rutile (110) Surface
BAO Bing1, SHEN Yue2, ZHANG Li-xian3, PAN Yong4, XU Yan-ting2, GUO Jun-mei2, WEN Ming2 *
(1. Sino-Platinum Metals (Shanghai) Co. Ltd., Shanghai 200050, China; 2. Kunming Institute of Precious Metals, Kunming 650106, China;3. The First People's Hospital of Yunnan Province, The Affiated Hospital of Kunming University of Science and Technology, Kunming 650032, China; 4. Southwest Petroleum University, Chengdu 610002, China;)
The first-principle method was used to study the interaction of Ag and rutile TiO2(110) surface. The adsorption of Ag on two types of bridges, O bridge or Ti bridge of rutile (110), was calculated respectively. It was found that the adsorption energy of Ag on Ti bridge was lower than that of O bridge. Further calculations on population, bond lengths and electronic structure showed that the bonding energy between Ti and Ag was larger on Ti bridge than that of O bridge, suggesting Ag atom was easier to form corresponding compounds with Ti atom on Ti bridge of rutile (110).
computational materials science; first-principle; Ag; rutile TiO2; bridge; electronic structure
O469;TG146.3
A
1004-0676(2020)02-0011-05
2019-05-07
国家自然科学基金(51564025、51004055);云南省基金重点项目(2017FA029)
鲍 冰,男,助理工程师,研究方向:贵金属新材料。E-mail:baobing@ipm.com.cn
闻 明,男,博士,研究员,研究方向:稀贵金属溅射靶材及表面科学。E-mail:wen@ipm.com.cn
