纯化前后C57BL/6小鼠骨髓细胞诱导破骨细胞的方法研究
2020-11-13杜天舒朱澍闫昭张大伟曹晓瑞
杜天舒,朱澍,闫昭,张大伟,曹晓瑞
(空军军医大学西京骨科医院关节外科,陕西 西安 710032)
破骨细胞(osteoclast,OC)被认为是体内唯一具有骨吸收功能的细胞类型,在骨代谢平衡中扮演着重要作用。破骨细胞过度活跃可导致多种骨疾病,如骨质疏松、关节假体松动、类风湿性关节炎、骨肿瘤、骨折后骨愈合延迟、Paget骨病等[1],因此对破骨细胞生成和功能影响的研究一直是领域内的热点重点。而破骨细胞体外培养中分离和纯化却长期困扰着研究者。现体外培养破骨细胞多利用原代细胞及单核/巨噬细胞Raw 264.7。相比细胞系,利用原代细胞诱导来反映破骨细胞活性有一定优势,但其体外诱导破骨细胞始终效果不佳,文献报道的诱导方法也不尽相同,无法有效套用。本研究通过研究纯化前后C57BL/6小鼠原代骨髓细胞向破骨细胞体外诱导的条件特点,总结利用原代细胞诱导技巧,提高诱导效率,为其体外研究提供方法学基础。
1 资料与方法
1.1 实验动物 8周龄SPF级C57BL/6雄性小鼠10只,由空军军医大学实验动物中心提供,体重在(20±2)g之间,健康状况良好。
1.2 试剂与材料 α-MEM培养基,青霉素/链霉素、红细胞裂解液(美国HyClone公司);胎牛血清(fetal bovine serum,FBS)(美国Gibco公司);小鼠骨髓单核细胞分离液试剂盒(北京索莱宝科技有限公司);抗小鼠CD11b荧光抗体(美国BD公司);牛血清蛋白(bull serum albumin,BSA)(德国Bioforxx公司);小鼠重组M-CSF(美国PeproTech公司);小鼠重组RANKL(美国R&D systems公司);TRAP染色试剂盒(美国Sigma公司);TRAP活性检测盒(南京建成生物工程研究所);逆转录试剂盒(日本TaKaRa公司);定量聚合酶链反应(quantitative polymerase chain reaction,q-PCR)引物(上海生工生物工程股份有限公司)。
1.3 实验仪器 流式细胞分析仪(美国BD公司);显微镜(日本Olympus公司)。
1.4 实验方法
1.4.1 小鼠骨髓来源原代细胞获取 脱颈处死小鼠,无菌条件下取股骨及胫骨,剔除骨质周围软组织,用含1%青霉素/链霉素的磷酸盐缓冲液(phosphate buffer saline,PBS)冲洗,剪除两侧干骺端,5 mL无菌注射器抽取无血清α-MEM培养基反复冲洗骨髓腔,直到髓腔内呈白色。1 000 rpm离心5 min,弃上清,加入红细胞裂解液,将细胞吹打混匀,37℃裂解10 min,再次1 000 rpm离心5 min,弃上清后用含10% FBS,1%青霉素/链霉素的α-MEM培养基重悬骨髓细胞,接种于25 cm2培养瓶中,37℃、体积分数5% CO2培育箱静置培养。
1.4.2 小鼠骨髓单核/巨噬细胞获取 收集过夜后骨髓原代细胞中未贴壁细胞,视为骨髓源性单核/巨噬细胞(bone marrow monocytes,BMMs),即破骨细胞前体细胞。1 000 rpm离心5 min,用无血清α-MEM培养基重悬细胞3次,离心后再用含10 %FBS,1%青霉素/链霉素的α-MEM培养基重悬细胞,镜下计数,分别以ρ=5×105cells/cm2及ρ=2.5×105cells/cm2接种于96孔板及6 cm皿内。
1.4.3 小鼠骨髓单核/巨噬细胞分离纯化 利用1.4.1中骨髓原代细胞按照小鼠骨髓单核细胞分离液试剂盒说明分离纯化出BMMs,以ρ=2.5×105cells/cm2接种于96孔板及6 cm皿内。
1.4.4 小鼠骨髓单核细胞表面抗原鉴定 将1.4.2及1.4.3中BMMs计数后取至少106个细胞收集于四个EP管(1、2、3、4管)中,离心后去除培养基。1管及3管分别加入1% BSA 100 μL冰上封闭30 min,作为阴性对照。2管及4管加入0.5 μL抗小鼠CD11b荧光抗体,避光冰上静置30 min。四管离心后分别加入PBS清洗,再加入适量多聚甲醛,转移至流式管后上机检测。
1.4.5 破骨细胞诱导分化 将1.4.2中铺板细胞设为4组(A、B、C、D组),A、B组细胞铺板ρ=5×105cells/cm2,其中A组于细胞铺板后即刻向96孔板及6 cm皿中加入20 ng/mL M-CSF及50 ng/mL RANKL,每2天换液,待第7天收样,96孔板内细胞行TRAP染色,6 cm皿中细胞提取mRNA行q-PCR检测。B组于细胞铺板后先用20 ng/mL M-CSF预处理3 d后,再一并加入50 ng/mL RANKL,之后每2天换液,待第7天收样,96孔板内细胞行TRAP染色,6 cm皿中细胞提取mRNA行q-PCR检测。C、D组细胞接种密度ρ=2.5×105cells/cm2,诱导因子M-CSF及RANKL的加入方法分别同A、B组。将1.2.3中铺板细胞设为E、F组,诱导因子M-CSF及RANKL的使用及诱导7 d后细胞收样方式分别同A、B组。
1.4.6 破骨细胞TRAP染色 根据TRAP染色试剂盒说明对A~F组96孔板中细胞行TRAP染色,核≥3个的TRAP阳性细胞视为成熟破骨细胞。
1.4.7 破骨细胞TRAP活性检测 根据TRAP活性试剂盒说明对A~F组6 cm皿中的细胞上清行TRAP活性检测。
1.4.8 破骨细胞标志性基因的q-PCR检测 将提取出的mRNA逆转录成单链DNA(cDNA),q-PCR检测破骨细胞标志性基因TRAP、组织蛋白酶(cathepsin K,CTSK)、活化T细胞核因子1(nuclear factor of activated T cells c1,NFATc1)、c-Fos的表达水平。目的基因引物序列见表1。

表1 q-PCR引物
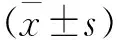
2 结 果
2.1 流式细胞分析结果 未纯化前小鼠骨髓原代细胞中CD11b阳性细胞比例约为(6±2)%,纯化后CD11b阳性细胞比例约为(58±3)%,提示利用单核细胞分离液纯化骨髓原代细胞后,其中单核细胞比例明显增高(见图1)。

a 未纯化前骨髓原代细胞 b 纯化后原代细胞
2.2 TRAP染色结果及定量分析 在M-CSF及RANKL的刺激下,细胞诱导第7天,各组细胞经TRAP染色结果各不相同。各组破骨细胞数量比较,B、C、D组几乎没有TRAP阳性细胞出现[B组(3.30±0.67)个/孔,C组(5.67±0.87)个/孔,D组(4.00±0.58)个/孔],提示无破骨细胞诱导生成。反之,与B、C、D组相比,A、E、F组中明显可见数量不一的TRAP阳性细胞[A组(257.30±8.35)个/孔,E组(285.70±7.62)个/孔,F组(373.00±19.56)个/孔]。镜下细胞多呈类圆形或不规则形,胞浆内酒红色TRAP阳性颗粒均匀沉淀,体积明显较单核细胞大数倍,可达50~100 μm,多核(核≥3个),周边可见伪足伸展,胞内可见囊泡。其中F组中TRAP阳性多核细胞数量及面积较其他组更明显(见图2~3)。
2.3 TRAP活性结果 将细胞诱导7 d后收集各组培养上清行TRAP活性检测,结果相比B、C、D三组[B组(10.00±1.16)U/L,C组(9.00±1.14)U/L,D组(6.67±1.45)U/L],A、E、F组的细胞上清中TRAP活性明显增高[(24.33±2.33)U/L,(28.67±2.03)U/L,(38.37±2.01)U/L)],差异有统计学意义(见图4),提示同TRAP染色结果相一致,A、E、F组中明显有大量破骨细胞生成。
2.4 q-PCR结果 相比B、C、D组,A、E、F组中破骨细胞分化相关标志性基因TRAP、CTSK、NFATc1及c-Fos的表达水平明显增高(见表2,见图5),差异有统计学意义,提示在A、E、F组中有大量BMMs分化成为破骨细胞。
3 讨 论
破骨细胞被认为是体内唯一具有骨吸收能力的细胞,其活性大小关系到整个机体的骨性质量[2]。破骨细胞活性过小可导致骨硬化病,而活性过大则会引起骨质疏松、骨折后骨愈合延迟、关节假体松动、骨坏死等骨质减少疾病[3],从而严重影响患者生活质量,因此研究者对影响破骨细胞活性(包括分化或功能)的研究从未停滞过。破骨细胞体外培养是研究破骨细胞活性的基础,但也是困扰研究者的难题。破骨细胞来源于造血干细胞谱系,是由多个单核/巨噬细胞融合而成,属于高代谢终末分化细胞,具有不能传代、生存时间短的特点[4-5]。利用原代细胞或单核/巨噬细胞RAW 264.7细胞系,通过诱导剂在体外诱导培养破骨细胞的诱导法,近年来被广泛用于破骨细胞的研究。细胞系诱导出来的破骨细胞纯度高,数量多,均一性好,但随着培养代数增加,细胞系会出现细胞生理性状丢失可能,故在体现细胞性状和细胞生物学特性方面远不及原代细胞。原代细胞拥有正常细胞形态,并且能够体现出细胞在生物体内所应具备的标记和功能,更接近生理状态,由此获得的数据具有更好相关性和更高价值。但利用原代细胞体外培养诱导分化破骨细胞往往难度较大,成功率不高,诱导质量不佳。Guo等[6]取培养48 h后未贴壁骨髓原代细胞,以ρ=5×105cells/mL接种,加之50 ng/mLM-CSF+100 ng/mL RANKL进行破骨细胞诱导;Kim等[7]取培养24 h后未贴壁骨髓原代细胞,30 ng/mL M-CSF预处理3 d后,以ρ=1×104cells/孔接种于96孔板,30 ng/mL M-CSF+100 ng/mL RANKL处理4 d以获得成熟破骨细胞;Feng等[8]用35 ng/mL M-CSF处理BMMs后以ρ=8×103cells/孔接种于96孔板,35 ng/mL M-CSF+50 ng/mL RANKL处理5~7 d;Kang等[9]在10 ng/mL M-CSF条件下培养骨髓原代细胞24 h后取未贴壁细胞,50 ng/mL M-CSF预处理3 d后,以ρ=1×104cells/孔接种于96孔板,30 ng/mL M-CSF+50 ng/mL RANKL处理3~5 d。可见文献中有关利用原代细胞体外诱导破骨细胞报道的方法不尽相同,单位不统一,难以单纯套用,还需研究者根据自身细胞特点摸索出合适的诱导方法,并且当使用不同规格培养板/皿配合不同实验时还需换算接种密度,费时费力且无法保证结果。因此,总结出便于研究者掌握的利用原代细胞体外诱导破骨细胞的特点和方法技巧,具有十分重要的意义和价值。

a A组 b B组

c C组 d D组
e E组 f F组

图3 TRAP阳性多核细胞数量化比较 图4 各组诱导7 d细胞上清TRAP活性

表2 各组破骨细胞分化相关标志性基因的表达水平

a TRAP b CTSK
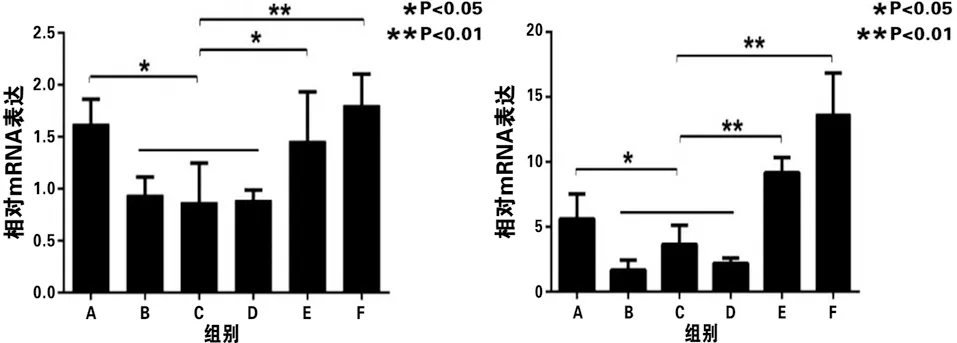
c NFATc1 d c-Fos
通过查阅文献,我们发现研究中关于破骨细胞体外诱导的接种密度各不相同,经过换算范围多集中在104 ~106 cells/cm2间,并且加入诱导剂M-CSF及RANKL的浓度及时机也不尽相同[6-10]。故我们按照不同接种密度(5×105cells/cm2和2.5×105cells/cm2)及诱导因子刺激方式(铺板时直接加入M-CSF和RANKL,和先加入M-CSF预刺激3 d后再一并加入RANKL)设计A~D四组实验。结果发现,单纯利用过夜贴壁法收集得来的BMMs,用较低密度接种(ρ=2.5×105cells/cm2),M-CSF & RANKL共刺激法和M-CSF预刺激法这两种方式均无法成功诱导出成熟破骨细胞(C、D组),当提高接种密度后(如ρ=5×105cells/cm2),相比M-CSF预刺激法,共刺激法可使BMMs成功分化为成熟破骨细胞。单核细胞纯度对于破骨细胞体外培养十分重要。通过测定,我们发现过夜后直接收集的细胞悬浮液中单核细胞纯度约为6%,细胞接种密度过低,细胞间距过大,故M-CSF和RANKL无法起到诱导作用,破骨细胞无法融合形成。加大细胞接种密度以增加单位内单核细胞数量,通过刺激单核/巨噬细胞可融合分化成破骨细胞。但不同于董伟[11]的研究,细胞经过M-CSF预刺激后再接受M-CSF和RANKL共刺激,仍没有形成破骨细胞。M-CSF可促进单核细胞向巨噬细胞和破骨细胞分化,RANKL是破骨细胞分化过程中不可缺少的诱导因子[12-13]。我们认为在单核细胞总数较少并伴有其他细胞(如淋巴细胞、内皮细胞等)混杂情况下,M-CSF会促使单核细胞贴壁并向成熟巨噬细胞方向分化,而后者几乎丧失再融合分化成破骨细胞能力,故导致M-CSF预刺激组培养效果不佳(B组)[14-15]。RANKL的早期加入不仅可以协同增加M-CSF促细胞增殖及存活能力,而且还能促使单核细胞向未成熟巨噬细胞方向分化,并进一步分化为破骨细胞(A组)[16-17]。当我们通过分离液纯化骨髓原代细胞,提高单核细胞纯度(E、F组)后发现,即使以较低密度铺板时,无论是何种诱导方式都能成功诱导出破骨细胞,且预刺激组诱导效果更佳。
综上所述,本实验通过对骨髓原代细胞的处理,BMMs接种密度以及诱导方式这几个方面进行探索研究,总结利用原代细胞体外诱导破骨细胞的一般特点,认为当细胞悬浮液中单核细胞比例不高时,应加大细胞接种密度以提高单核细胞数量,并在M-CSF刺激下尽早加入RANKL,促进单核细胞向破骨细胞分化;当保证单位内单核细胞数量前体下,可适当降低细胞接种密度以减少细胞间接触抑制影响,并可利用M-CSF预处理后再行与RANKL共诱导,从而提高破骨细胞数量和质量,为下一步体外研究破骨细胞特点打下基础。
