含氟菲咯啉的设计合成及其杂配铜配合物的光解水性能
2020-11-11刘雪粉俞哲健徐良轩王天琦罗书平
刘雪粉 俞哲健 徐良轩 陈 浩 王天琦 杨 鹏*, 罗书平*,
(1杭州师范大学钱江学院,杭州 310018)
(2浙江工业大学绿色化学合成技术国家重点实验室培育基地,杭州 310014)
太阳能作为一种清洁无污染的可再生能源,具有体量大、易得等优势,利用太阳能直接分解水使其光能转换成氢气化学能,是当今新能源的研究前沿,也是解决能源问题和环境问题的重要途径之一[1]。目前人工光解水制氢系统是由捕光发光体或光敏剂(photosensitizers,简称PS)、水还原催化剂(water reduction catalysts,简称 WRC)、牺牲剂(sacrificial reductant,简称 SR)组成[2],其中 PS的作用是吸收光子并将能量传递给那些不能吸收光子的催化分子。然而目前PS是制约太阳能利用效率的关键问题,因此研究发展高效、稳定、价廉、无毒的PS(或光能转换材料)是光解水体系中的重要研究任务。1970年[Ru(bpy)3]2+首次被用作PS[3],随后其他Ru配合物[4]和一系列其他贵金属配合物,如 Rh[5]、Pt[6]、Ir[7]等都成功运用在光催化制氢中,然而价格昂贵、储量稀少、环境污染的缺点限制了它们的使用。在过去40年间,非金属有机染料[8]、金属配合物[9]和官能化金属-有机骨架材料[10]都已被广泛用作光催化制氢的PS研究中。由于金属与其配体之间的强偶合作用,金属配合物具有更高的稳定性[11]。因此,开发高效的金属配合物PS在该领域有很好的前景。
在光催化体系中,PS能有效地吸收入射光子并将其转换成激发态[12]。由于氟化合物具有高效的电子传输效率,近年来,众多科研人员在有机光电材料中引入氟原子用以调节光电材料的光电性质[13]。从2012年起,我们着重研究了非贵金属的氮磷杂配铜配合物作为高效稳定的PS,并探究了1,10-菲咯啉2,9位和4,7位的修饰对铜基光敏剂光解水制氢活性以及光电物理性能的影响,研究发现2,9位为含氟的菲咯啉氮配体,其有利于提升氮磷杂配铜配合物的光敏性能[14]。
我们尝试在菲咯啉4,7位引入含氟原子的取代基,并进一步研究其作为氮配体对铜配合物光敏活性的影响,通过结构设计与合成方法的探索,我们成功制备了一种新型的4,7-含氟芳基取代菲咯啉衍生物(图1)。在此基础上,着重研究了氟原子的氟化效应对杂配铜光敏剂在光解水中制氢活性的影响,并且对其光电物理性质进行探讨。

图1 4,7-二氟芳基-2,9-二甲基-1,10-菲咯啉衍生物的合成路线Scheme 1 Synthesis route of 4,7-bis(fluoroaryl)-2,9-dimethyl-1,10-phenanthroline derivatives
1 实验部分
1.1 仪器与试剂
原料及试剂均购自商业公司,所有溶剂都经过重蒸除水;NMR分析使用瑞士Bruker公司的AdvanceⅢ核磁共振仪;紫外分析使用瓦里安公司的CARY100紫外-可见-近红外分光光度计;荧光分析使用SHIMADZU公司的RF-6000荧光分析仪;循环伏安(CV)分析使用上海辰华的CHI电化学工作站;质谱(MS)分析使用美国Thermo公司LCQ advantage质谱仪;高分辨质谱(HR-MS)分析使用美国Agilent公司的Agilent 6210 TOF LC/MS。
1.2 合 成
1.2.1 4,7-二(氟芳基)-2,9-二甲基-1,10-菲咯啉衍生物的合成
4,7-二(氟芳基)-2,9-二甲基-1,10-菲咯啉衍生物的合成路线参见图1。
1.2.2 中间体SM-1和中间体SM-2的合成
按参考文献[15]合成。
1.2.3 不同含氟芳基取代基的1,10-菲咯啉配体的合成
1.2.3.1 4,7-二溴-2,9-二甲基-1,10-菲咯啉(SM-3)的合成[16]
在氩气保护下,向三口烧瓶中依次加入中间体SM-2(2.93 g,11.65 mmol)和200 mL三溴氧磷,升温至回流并反应6 h。原料SM-2反应过程由薄层色谱(TLC)监测,反应结束后冷却至室温。将反应液减压脱溶后,缓慢倒入冰水混合物中,剧烈搅拌,然后再用1 mol·L-1的氢氧化钠水溶液调至pH=10左右,有固体析出。用二氯甲烷萃取3次,无水硫酸钠干燥,硅胶柱层析纯化(二氯甲烷和乙酸乙酯体积比为20∶1)得到2.74 g白色粉末(SM-3),收率:70%;1H NMR(500 MHz,CDCl3):δ8.17(s,2H),7.83(s,2H),2.92(s,6H);13C NMR(125 MHz,CDCl3):δ159.81,145.59,134.30,127.97,126.34,124.96,25.49。
1.2.3.2 4,7-二(4-氟苯基)-2,9-二甲基-1,10-菲咯啉(N1)的合成
在氩气保护下,向Schlenk管中依次加入中间体SM-3(0.365 g,1 mmol)、4-氟苯硼酸(0.35 g,2.5 mmol)、碳酸钾(1.10 g,8 mmol)、四(三苯基膦)钯(0.115 g,0.1 mmol)、3 mL甲苯、3 mL乙醇和3 mL去离子水,逐渐升温至80℃并搅拌24 h。TLC监测原料SM-3反应完后,用硅藻土过滤,并用二氯甲烷洗涤滤饼,合并有机相,采用无水硫酸钠干燥,硅胶柱层析纯化(二氯甲烷和乙酸乙酯体积比为30∶1)得到0.317 g白色粉末(N1),收率:80%;1H NMR(500 MHz,CDCl3):δ7.72(s,2H),7.53~7.46(m,4H),7.44(s,2H),7.25~7.17(m,4H),3.00(s,6H);13C NMR(125 MHz,CDCl3):δ162.98(d,J=248.5 Hz),158.98,147.73,145.83,134.15(d,J=3.4 Hz),131.48,131.41,124.79,124.22,122.95,115.82,115.65,25.94;19F NMR(375 MHz,CDCl3):δ-113.19;ESI-HRMS:m/z:Calcd.for C26H19F2N2:397.151 1[M+H]+;Found:397.153 0[M+H]+。
1.2.3.3 4,7-二(3,5-二氟苯基)-2,9-二甲基-1,10-菲咯啉(N2)的合成
合成方法参照N1。
化合物N2:黄色固体,收率78%;1H NMR(500 MHz,CDCl3):δ7.73(s,2H),7.44(s,2H),7.09~7.01(m,4H),6.94(tt,J=8.9,2.3 Hz,2H),2.99(s,6H);13C NMR(125 MHz,CDCl3):δ164.08(d,J=12.9 Hz),162.09(d,J=12.8 Hz),159.32,146.36,145.93,141.29(t,J=9.5 Hz),124.29,123.97,122.94,113.08,112.77,104.06(t,J=25.1 Hz),26.03;19F NMR(375 MHz,CDCl3):δ-108.69;ESI-HRMS:m/z:Calcd.for C26H17F4N2:433.432 2[M+H]+;Found:433.134 0[M+H]+。
1.2.3.4 4,7-二(4-(三氟甲基)苯基)-2,9-二甲基-1,10-菲咯啉(N3)的合成在氩气保护下,向三口烧瓶中依次加入中间体SM-3(0.5 g,1.37 mmol)、4-(三氟甲基)苯硼酸(0.86 g,4.5 mmol)、碳酸钾(1.02 g,7.4 mmol)、醋酸钯(0.01 g,0.05 mmol)、三环己基膦(0.03 g,0.1 mmol)、10 mL 1,4-二氧六环和2 mL去离子水,逐渐升温至回流并搅拌24 h。采用TLC监测,原料反应完后,用硅藻土过滤,并用二氯甲烷洗涤滤饼合并有机相,无水硫酸钠干燥,硅胶柱层析纯化(二氯甲烷和乙酸乙酯体积比为20∶1)得到0.584 g白色粉末(N3),收率:86%;1H NMR(500 MHz,CDCl3):δ7.80(d,J=8.0 Hz,4H),7.70~7.62(m,6H),7.47(s,2H),3.02(s,6H);13C NMR(125 MHz,CDCl3):δ159.11,147.19,145.76,141.70,130.70(d,J=32.9 Hz),125.62,125.57(q,J=3.8 Hz),125.53,125.09,124.40,124.05,122.93,122.88,25.79;19F NMR(375 MHz,CDCl3):δ-62.52;ESI-HRMS:m/z:Calcd.for C28H19F6N2:497.144 7[M+H]+;Found:497.147 0[M+H]+。
1.2.3.5 4,7-二(3,5-二(三氟甲基)苯基)-2,9-二甲基-1,10-菲咯啉(N4)的合成
合成方法参照N1。
化合物N4:黄色固体,收率82%;1H NMR(500 MHz,CDCl3):δ8.03(d,J=1.8 Hz,2H),7.99(d,J=1.7 Hz,4H),7.63(s,2H),7.52(s,2H),3.04(s,6H);13C NMR(125 MHz,CDCl3):δ159.80,145.89,145.69,140.18,132.47(q,J=33.7 Hz),129.83(d,J=3.9 Hz),126.47,124.62,124.32(d,J=3.5 Hz),122.91,122.79,122.44,122.13,119.96,25.98;19F NMR(375 MHz,CDCl3):δ-62.71;ESI-HRMS:m/z:Calcd.for C30H17F12N2:633.119 5[M+H]+;Found:633.122 3[M+H]+。
1.2.4 铜配合物的原位合成
参考课题组之前的工作[17],采用原位配位的方法得到相应的铜配合物。以铜配合物CP1为例:在氩气保护下,将磷配体Xantphos(1 mmol)和六氟磷酸四乙腈合铜(1 mmol)溶于20 mL二氯甲烷中搅拌5 min,再加入氮配体N1(1 mmol),室温下搅拌2 h,得到相应的氮磷杂配铜配合物CP1(图2),浓缩后即可用于光谱测试和CV测试。
1.3 光解水制氢测试
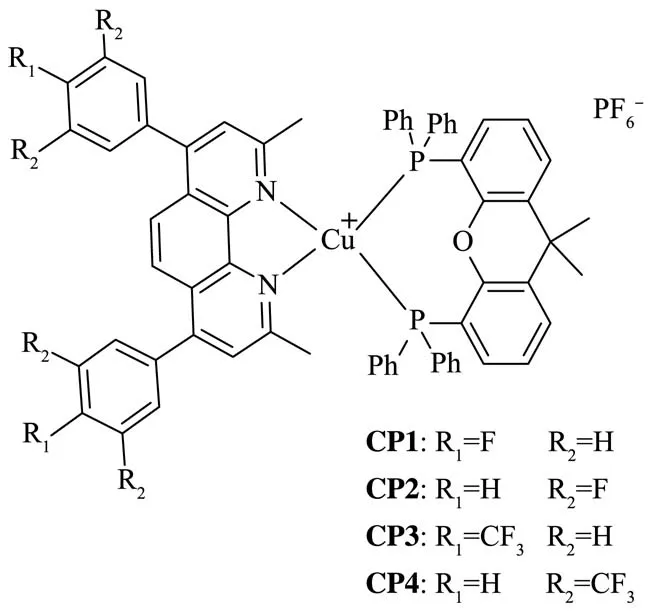
图2 铜配合物CP1~CP4的结构Scheme 2 Structure of copper complexes CP1~CP4
以氮配体N1为例:在氩气保护下,将磷配体Xantphos(3.5 μmol)、六氟磷酸四乙腈合铜(3.5 μmol)和 10 mL混合溶剂(VTHF∶VTEA∶VH2O=4∶3∶1,四氢呋喃(THF)为溶剂,三乙胺(TEA)为牺牲剂)加入反应瓶中,用循环恒温水浴槽将反应体系温度稳定在25 ℃,搅拌 15 min。再加入 N1(3.5 μmol),搅拌 5 min,最后加入催化剂 PdCl2和 Xantphos(各 5 μmol),打开氙灯光源照射反应瓶,并用U形管收集产生的氢气,记录产生气体的体积。待反应体系中不产生气体时,停止搅拌,关闭光源,并用气相色谱检测收集的气体。
1.4 光谱测试
分别将1 mmol原位合成的CP1~CP4铜基配合物(1 mmol)用THF溶解后定容到50 mL容量瓶中,在室温下进行光化学测试。
1.5 CV测试
配制0.1 mol·L-1四丁基六氟磷酸铵的THF电解质溶液,加入3.5 μmol的铜基配合物,定容到100 mL容量瓶中,在氩气条件下,用三电极体系(以d=2 mm的玻碳电极为工作电极,以铂为对电极,以Ag/AgNO3为参比电极)在室温下进行CV测试,电解质是0.1 mol·L-1四丁基六氟磷酸铵的THF溶液。CV实验条件:扫描速度100 mV·s-1,阶跃电位2.5 mV,调制振幅25 mV,调制时间0.05 s,间隔时间0.5 s,最高电位1.8 V,最低电位-3.0 V。
2 结果与讨论
2.1 紫外和荧光表征
从图3可知,铜配合物CP1~CP4在400 nm左右的紫外吸收峰是铜基光敏剂的特征吸收峰,该吸收峰归因于金属-配体的电子转移(MLCT)[18]。由于[Cu(P^P)(N^N)]+配合物在溶液中会产生配体之间的重组,导致氮配体与铜离子形成均配的[Cu(N^N)2]+配合物,所以CP1在480 nm左右的紫外吸收中有一个比较小的特征吸收峰。而随着F原子数目不断增加,铜基光敏剂在最大吸收波长处红移,摩尔吸光系数(ɛ)明显下降,说明F原子的强吸电子效应能够显著拉低氮配体的电子云密度,降低MLCT的能垒。荧光发射光谱表明(图3和表1),4种铜配合物CP1~CP4在480~700 nm之间的光谱发射带很宽并且没有明显的电子振动序列,进一步证明该PS在激发态下具有显著的电荷转移特性。通过比较铜配合物CP1~CP4的最大发射波长(λmax)和荧光量子效率(Φabs),发现CP1的λmax最小(564 nm),荧光量子效率最高(达到0.43)。随着F原子的增加,其最大发射波长发生红移,且ɛ也随之减少,可能原因是随着F原子数量的增加,杂配铜光敏剂的生成得到抑制,进而使得不同PS的ɛ存在较大差异。对比铜配合物CP4,CP1的ɛ和Φabs均最大,因此其具有最好的制氢活性。

图3 CP1~CP4的紫外吸收(左边)和荧光发射光谱(右边)Fig.3 Absorption(left)and emission(right)spectra for CP1~CP4
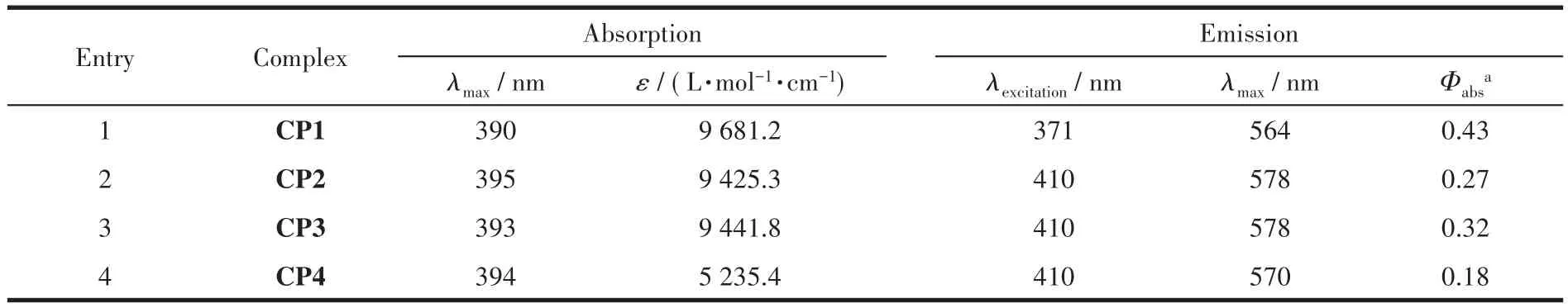
表1 CP1~CP4的电子吸收和发射光谱数据Table 1 Electronic absorption and emission spectrum data for CP1~CP4
2.2 循环伏安测试
由图4可知,铜配合物CP1~CP4均有相似的氧化还原电位(Eoxd=-0.6 V,Ered=-1.6 V),主要归属于菲咯啉配体的氧化还原[19]。就循环体系而言,整个过程是一个单电子还原过程。4种配合物的氧化还原电位相似,这可能是由于配合物的整体骨架不变,简单的修饰无法大幅改变其光物理性能。

图4 CP1~CP4的CV曲线Fig.4 CV curves for CP1~CP4
2.3 制氢性能研究
由图5可知,当使用氮配体N1时该体系获得最高活性,产氢38.4 mL,即CP1的TON高达896,这与紫外吸收和荧光发射光谱结果相符。

图5 CP1~CP4的光解水结果Fig.5 Results for the photocatalytic water spliting using CP1~CP4
在之前工作[20]的基础上,我们首先以配合物CP1作为研究对象研究了牺牲剂的种类对于光解水制氢的影响(表2)。结果表明,带有Xantphos和这些新型氮配体的铜配合物应用于光解水时表现出显著的光催化活性。选用铜光敏剂经常匹配的Fe3(CO)12为催化剂时[17a],TEA为最佳的电子牺牲剂,制氢总转换数达289。由于制氢催化剂的种类对光解水制氢活性影响很大,我们继续探索了该体系催化剂的种类对其性能的影响。从表3可以看出,当K2PtCl4作为催化剂时,催化活性不理想,而PdCl2作为催化剂时具有良好的催化活性。之后,用磷配体与PdCl2的配合物作为催化剂探究其制氢性能。对比PdCl2+PCy3、PdCl2+Xantphos和[Ru(bpy)3]2可知,磷配体刚性越强,催化活性越高,这归因于刚性强的磷配体有利于提高催化剂的稳定性,其中当Xantphos为磷配体时,TON达到了896。同样条件下,将常用的贵金属光敏剂[Ru(bpy)3]2应用于该制氢体系中时,仅产生了2.5 mL氢气,TON远低于铜光敏剂CP1。氟原子的不断引入导致PS的激发态寿命不断减少,不利于激发态到催化剂之间的电子转移,这使得整个制氢体系的稳定性随之下降。
增加磷配体的含量不会提高光解水制氢的活性(表4),反而会降低其活性,因为PdCl2很容易与2个磷形成配位键,而其余没有配位的磷与氮配体竞争铜配合物的配位点,不利于PS的稳定性。改变催化剂浓度对光解水制氢体系活性也有很大影响(表5)。当催化剂投入量从 1 μmol增大到 20 μmol时,TON先增大后变小,其中催化剂量为5 μmol时,最大TON为896。催化剂浓度过高会引起PS的中毒,催化剂浓度过低会使体系稳定性下降,二者均会减弱催化产氢的效果,所以5 μmol催化剂为最佳投料量。综上所述,最终建立的光催化体系是以CP1为铜基光敏剂(3.5 μmol),PdCl2和Xantphos为催化剂(5 μmol,物质的量之比为1∶1),TEA为电子牺牲剂,溶剂为10 mL体积比4∶3∶1的THF、TEA、H2O的混合溶剂。

表2 牺牲剂对光解水产氢的影响Table 2 Effect of SR on hydrogen production from water splitting

表3 催化剂种类对光解水制氢的影响Table 3 Effect of WRC on hydrogen production from water splitting
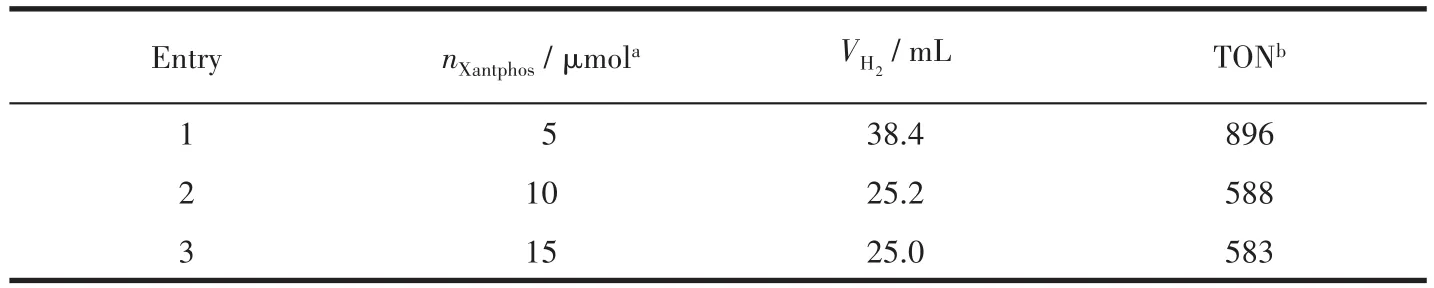
表4 磷配体含量对光解水产氢的影响Table 4 Effect of P ligand content on hydrogen production from water splitting

表5 催化剂用量对光解水产氢的影响Table 5 Effect of WRC content on hydrogen production from water splitting
为了确定在光解水制氢体系中铜基光敏剂激发态的淬灭途径,我们进行了一系列荧光淬灭测试(图 6)。测试过程如下:称取 3.5 μmol CP1 溶解在50 mL THF中,然后分别加入不同浓度的WRC(PdCl2和Xantphos的物质的量之比为1∶1)和TEA,在室温条件下测试体系的荧光强度。使用TEA作为淬灭剂,发现TEA的浓度对CP1的荧光强度几乎无影响。当WRC作为淬灭剂时,随着淬灭剂浓度的增大,荧光强度明显下降,原因可能是加入的WRC与铜基光敏剂的激发态之间发生了分子间的能量转移,说明该光解水制氢体系的一般机理可能为氧化淬灭径。
结合相关文献[21]以及上述淬灭实验数据,推测其光催化制氢机理如图7所示。在光照条件下,PS接受光子从基态激发达到激发态(PS*),然后与钯催化剂的活性中间体Pd(P^P)(OH)2发生电子迁移后生成PS+,中间体PS+再从牺牲剂TEA上获得一个电子后回到基态,进入下一个循环。生成的钯氢物种再通过另一个光敏循环后获得一个电子,接受溶液中的H+生成钯二氢物种,随后放出了一分子氢气,完成催化循环。
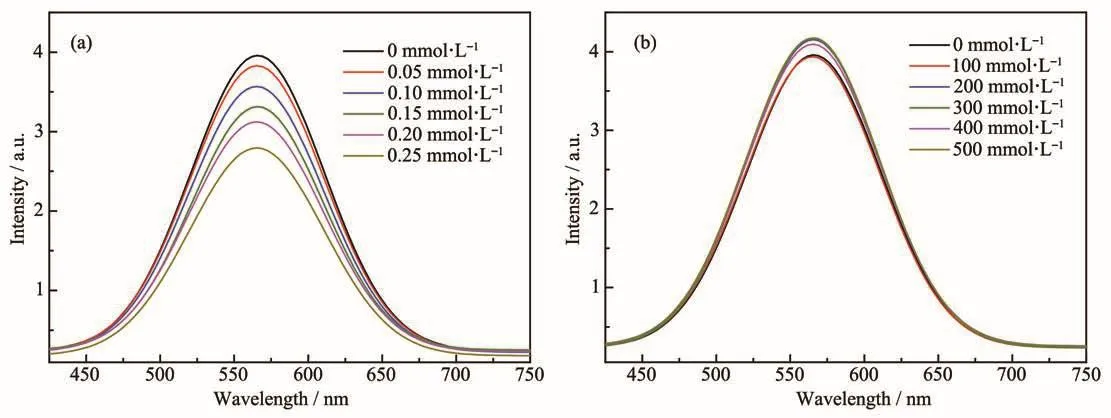
图6 WRC(a)和TEA(b)浓度对CP1的荧光淬灭Fig.6 Fluorescence spectra of CP1 quenched by different concentrations of WRC(a)and TEA(b)

图7 光解水制氢可能的反应机理Fig.7 Proposed reaction mechanism for the hydrogen production from water splitting
3 结论
设计合成了一系列4,7位含氟取代基菲咯啉氮配体(N1~N4),然后与六氟磷酸四乙腈合铜和二磷配体Xantphos通过原位配位形成铜基光敏剂CP1~CP4。光敏活性结果表明,CP1光解水制氢活性最高,产氢TON可达896。铜基光敏剂的光电物理性质测试结果表明4个配合物在溶液中具有很好的稳定性,不会发生配体的重组现象。通过荧光淬灭实验表明,氧化淬灭为主要的淬灭途径。
