近红外光谱法快速测定柴胡提取过程中的药效成分
2020-11-09杨留长纪晓亮李易航徐倩茹佟海滨
杨 越,杨留长,纪晓亮,李易航,徐倩茹,佟海滨*
(1.温州大学 生命与环境科学学院,浙江 温州 325035;2.浙江省水环境与海洋生物资源保护重点实验室,浙江 温州 325035;3.温州医科大学 公共卫生与管理学院,浙江 温州 325035)
提取是中药生产的首要工艺,其中间体的质量会直接影响后续生产过程的中间体及最终产品的质量。目前,提取工艺的质量控制主要采用抽样和离线方式,依据质量标准采用常规分析方法(如紫外-可见分光光度法、高效液相色谱法、气相色谱-质谱联用法)检测提取过程中间体。然而,这些方法费时费力,难以快速测定,无法实时监控生产过程中药效成分含量的变化情况,从而导致产品批次间的质量差异较大。因此,亟需研究一种适用于中药生产过程的快速分析方法,这将有助于实现中药生产过程的实时检测与优化控制,对于推进中药产业智能制造和保证产品质量具有重要的现实意义。
近红外光谱(NIRS)技术是一种间接的分析方法,往往需要结合计量学模型进行分析测定。其具有快速、无损、绿色,以及多组分同时测定等特点[1-4],目前已被广泛应用于中药药效成分的含量测定[5-6]、天然产物的鉴别[7-9]和药材的快速鉴定[10-12]等方面。此外,NIRS由于无需对样品进行预处理,特别适合在线分析,因而被越来越多地应用于中药生产过程的质量控制[13]。
柴胡化学成分复杂,其中皂苷类、黄酮类、多糖类为其主要活性成分[14-15],具有抗辐射、抗病毒、抗溃疡、降血脂、增强免疫力等疗效[16-17]。本文将NIRS应用于柴胡的提取过程研究,以总黄酮、多糖、柴胡皂苷A和柴胡皂苷D为质量指标,收集提取液样品,采集所有样品的近红外光谱图并采用传统分析方法测定其指标含量,结合偏最小二乘法(PLS)建立柴胡提取过程中各指标的定量校正模型,对各指标成分含量进行快速分析,以实现中药提取过程中的质量快速监测。
1 实验部分
1.1 仪器与材料
Agilent 1260高效液相色谱仪(Agilent Technologies,USA);Waters Xbridge C18色谱柱(4.6 mm×250 mm,5 μm);Antaris Ⅱ傅里叶变换近红外光谱仪(Thermo Fisher,USA);TU1810紫外可见分光光度计(北京普析通用仪器有限责任公司);FA2004电子天平(上海越平科学仪器有限公司);DK-S24电热恒温水浴锅(上海精宏实验设备有限公司);2 L三口圆底烧瓶。
聚醚砜超滤膜(直径:47 mm,孔径:0.22 μm,北京京启华诚科技发展有限公司);乙腈(色谱纯,Merck,Germany);浓硫酸、甲醇、苯酚、亚硝酸钠、氢氧化钠、硝酸铝(分析纯,国药集团化学试剂有限公司);芦丁、葡萄糖、柴胡皂苷A、柴胡皂苷D对照品(纯度均大于98%,成都曼思特生物科技有限公司);超纯水(Millipore,USA);实验用水均为超纯水;自药店购买藏柴胡,经温州大学生命与环境科学学院佟海滨副研究员鉴定。
1.2 提取液样品的制备
称取1 kg柴胡药材,置于2 L三口圆底烧瓶中,加入10倍量的水,煎煮60 min。煎煮自0 min开始每隔4 min收集10 mL提取液,并补充10 mL水,直至60 min为止。每批次煎煮60 min之后,立即连续收集5个提取终点样本。重复6次提取实验,共收集126个样品。
1.3 近红外光谱的采集
采集柴胡提取液样品的透射光谱,设定光谱扫描范围为4 000~10 000 cm-1,扫描次数为64次,分辨率为8 cm-1,液体样品池为0.5 mm光程的石英比色皿,以空气为参比,每份样品重复测定3次,取平均光谱用于后续数据处理以减小误差。
1.4 药效成分含量的测定
1.4.1 总黄酮含量的测定
(1)对照品溶液的制备; 采用芦丁作为对照品,利用紫外可见分光光度法测定柴胡中的总黄酮含量[18-20]。精密称定干燥至恒重的芦丁对照品10.12 mg,置于25 mL容量瓶中,加水适量,水浴微热使之溶解,加水至刻度,摇匀,即得质量浓度为404.8 μg/mL的芦丁对照品溶液。
(2)标准曲线的绘制; 精密量取芦丁对照品溶液0.0、0.5、1.0、1.5、2.0、2.5、3.0 mL,分别置于25 mL量瓶中,各加水至6.0 mL,加5%亚硝酸钠溶液1 mL,混匀,放置6 min,加10%硝酸铝溶液l mL,摇匀,放置6 min,加4%氢氧化钠试液10 mL,再加水至刻度,摇匀,放置15 min。以相应的试剂为空白对照,在500 nm波长处测定吸光度,以吸光度(Y)为纵坐标,芦丁的质量浓度(X,μg/mL)为横坐标绘制标准曲线。回归方程为Y=12.471X-0.000 04(0~48.576 μg/mL),r2为0.999 7,说明方法的线性关系良好。
(3)供试品溶液的制备; 取柴胡提取液于离心机中,3 800 r/min离心10 min后,取上清液3 mL于25 mL试管中,加水至6 mL,加5%亚硝酸钠溶液1 mL,混匀,放置6 min后,加10%硝酸铝溶液l mL,摇匀,放置6 min,加4%氢氧化钠试液10 mL,再加水至刻度,摇匀,放置15 min。以相应的试剂为空白对照,在500 nm波长处测定吸光度。
1.4.2 多糖含量的测定
(1)对照品溶液的制备; 采用无水葡萄糖作为对照品,利用紫外可见分光光度法测定柴胡中的多糖含量[20-22]。精确称定干燥至恒重的葡萄糖对照品4.00 mg,置于50 mL容量瓶中,加水适量,水浴微热使之溶解,加水至刻度,摇匀,即得质量浓度为0.080 mg/mL的葡萄糖对照品溶液。
(2)标准曲线的绘制; 精密量取葡萄糖对照品溶液0.0、0.2、0.4、0.6、0.8、1.0 mL,分别置于试管中,每管加入水使溶液总量达到1 mL。再分别加入6%苯酚0.5 mL和浓硫酸2.5 mL,振荡均匀,放置30 min。以相应试剂为空白对照,在490 nm波长处测定吸光度,以吸光度(Y)为纵坐标,葡萄糖的质量浓度(X,mg/mL)为横坐标绘制标准曲线。回归方程为Y=60.557X+0.000 8(0~0.020 mg/mL),r2为0.999 8,说明方法的线性关系良好。
(3)供试品溶液的制备;取柴胡提取液于离心机中,3 800 r/min离心10 min后,取上清液1 mL,置于250 mL容量瓶中定容至刻度,摇匀,精密量取1 mL,再分别加入6%苯酚0.5 mL和浓硫酸2.5 mL,振荡均匀,放置30 min。以相应的试剂为空白对照,在490 nm波长处测定吸光度。
1.4.3 柴胡皂苷A与柴胡皂苷D含量的测定
(1)对照品溶液的制备;取柴胡皂苷A、柴胡皂苷D对照品各适量,精密称定,置于10 mL容量瓶中,加甲醇溶解并定容得柴胡皂苷A、柴胡皂苷D标准溶液,各取柴胡皂苷A、D标准溶液1 mL置于10 mL容量瓶中,加甲醇至刻度,制成质量浓度分别为58、59 μg/mL的柴胡皂苷A与柴胡皂苷D混合对照品溶液。
(2)供试品溶液制备; 取柴胡提取液,用聚醚砜超滤膜过滤,弃去初滤液,取续滤液作为供试品溶液。
(3)HPLC条件;色谱柱:Waters Xbridge C18柱(4.6 mm×250 mm,5 μm);流动相:水-乙腈;检测波长:210 nm;进样量:10 μL;流速:1.0 mL/min;柱温:25 ℃;柴胡皂苷A梯度洗脱:0~20 min:45%~50%乙腈;柴胡皂苷D梯度洗脱:0~20 min:50%~58%乙腈。
(4)标准曲线的绘制;取柴胡皂苷A与柴胡皂苷D混合对照品溶液,按上述色谱条件分别进样1、2、3、4、5、6 μL,以峰面积(Y)为纵坐标,柴胡皂苷A的质量浓度(X,μg/mL)为横坐标绘制标准曲线。柴胡皂苷A的回归方程为Y=20.689X-0.862(5.8~34.8 μg/mL),r2为0.999 6,说明方法线性关系良好。同样取混合对照品溶液,按上述色谱条件分别进样0.8、1.2、1.4、1.6、1.8、2.0 μL,以峰面积(Y)为纵坐标,柴胡皂苷D的质量浓度(X,μg/mL)为横坐标绘制标准曲线。柴胡皂苷D的回归方程为Y=19.326X+5.214 5(4.72~11.8 μg/mL),r2为0.999 2,说明方法线性关系良好。
1.4.4 方法学考察
(1)精密度试验; 取一批样品溶液,在上述方法条件下连续测定6次,以总黄酮、多糖、柴胡皂苷A及柴胡皂苷D的吸光度或峰面积进行精密度考察。结果显示,各成分的相对标准偏差(RSD)分别为0.201%、0.084 8%、0.049%、0.442%,表明该方法的精密度良好,符合定量要求。
(2)稳定性试验; 取同一样品溶液适量,分别在0、1、2、4、8、12 h 6个时间点测定4种成分的吸光度或峰面积,进行稳定性考察。结果显示,各成分的RSD分别为0.683%、0.070%、0.210%、0.852%,表明样品在12 h内稳定性良好。
(3)重复性试验; 平行制备6份样品溶液,在上述方法条件下测定,计算总黄酮、多糖、柴胡皂苷A及柴胡皂苷D含量,进行重复性考察。结果显示,各成分的RSD分别为0.429%、0.402%、0.952%、2.470%,表明方法的重复性良好,符合定量要求。
1.5 模型性能评价及数据处理
模型性能的评价通常采用校正集误差均方根(Root mean square error of calibration,RMSEC)、预测集误差均方根(Root mean square error of prediction,RMSEP)、校正集相关系数(Correlation coefficient of calibration,RC)、预测集相关系数(Correlation coefficient of prediction,RP)、预测集相对偏差(Relative standard of errors of prediction,RSEP)作为评价指标,考察不同建模方法的优劣[23-24]。通常R越接近于1,RMSEC和RMSEP之间的差距越小,则代表模型的预测性能越高。选择合适的波段,在适宜的光谱预处理基础上,运用PLS建立NIRS与柴胡各指标含量测定值之间的定量校正模型。各性能评价指标的计算公式如下:
其中,Ci为参考方法测量值;i为NIRS模型预测值;Cn为校正集中Ci的平均值;Cm为预测集中Ci的平均值;n为校正集样本数;m为预测集样本数。
2 结果与讨论
2.1 质控指标测定结果
柴胡提取过程中的总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的动态变化曲线见图1。从图1中可以看出,提取液中4种药效指标的含量均随时间呈上升趋势。其中,总黄酮与多糖的含量在整个提取过程中持续上升;柴胡皂苷A含量呈现先快速增长再缓慢增长的趋势;柴胡皂苷D含量在前10 min呈现增长趋势,之后趋于稳定。
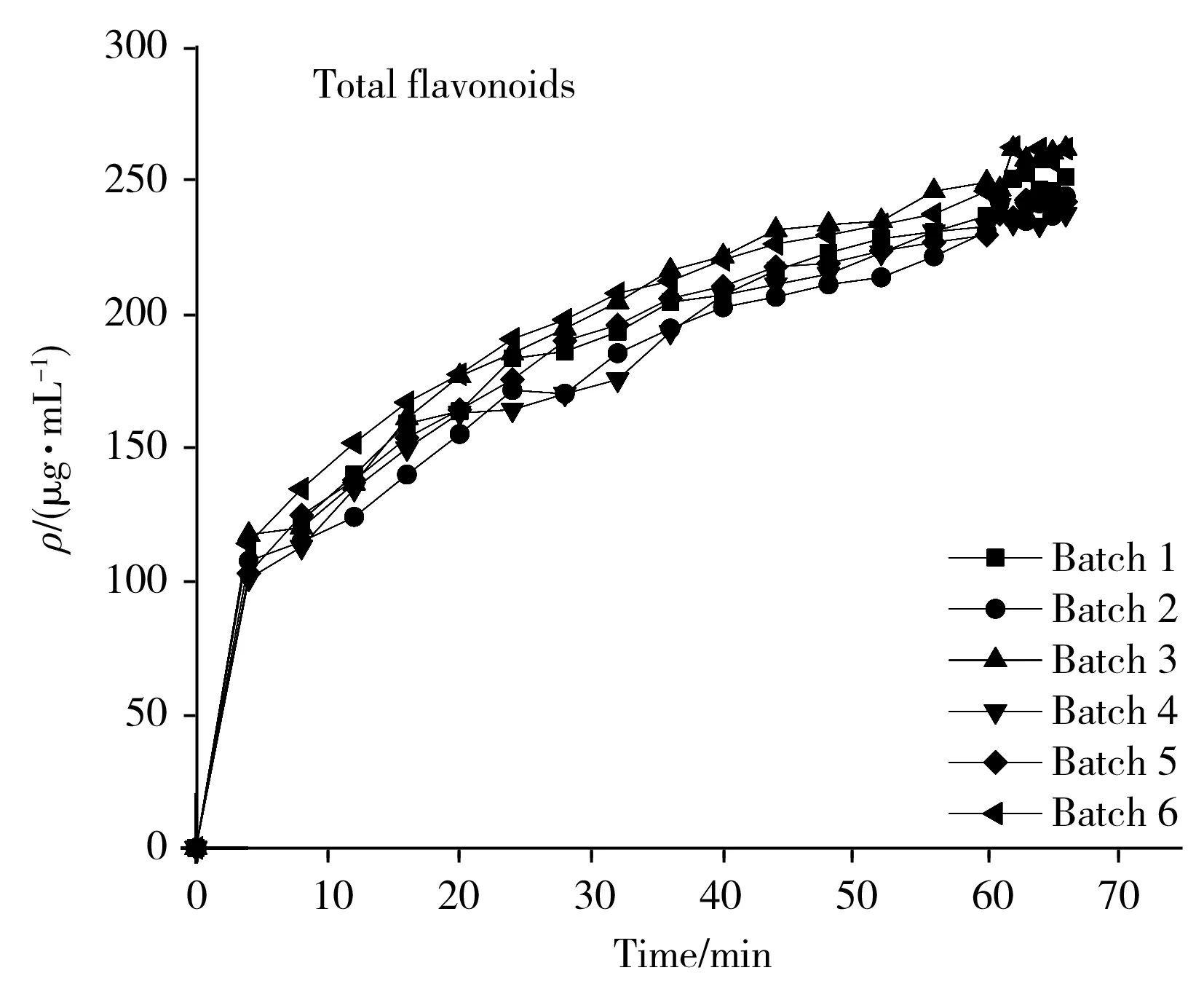
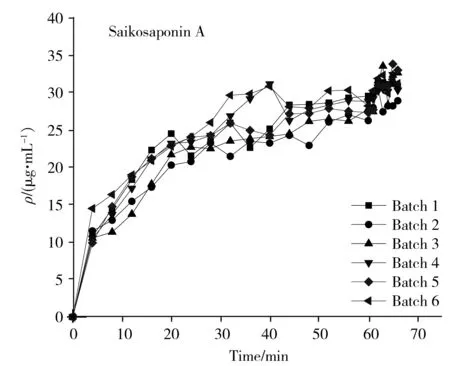
将第5批柴胡提取液样品作为预测集,剩余5个批次的样品作为校正集。各指标成分的含量分布如表1所示。可以看到,校正集中总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的含量范围分别为101.00~262.77 μg/mL、2.99~10.87 mg/mL、10.53~33.60 μg/mL、3.07~6.80 μg/mL;预测集中总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的含量范围分别为124.77~245.60 μg/mL、6.25~10.39 mg/mL、14.79~33.89 μg/mL、3.97~7.04 μg/mL。校正集指标成分的含量范围基本涵盖了预测集,表明样品分布合理,有利于建立高精度模型。

表1 校正集和预测集数据的统计结果Table 1 Statistical results of samples in the calibration set and prediction sets
2.2 近红外光谱的采集
近红外光谱区域的主要光谱信息来源于含氢基团如—OH、—NH和—CH 等的倍频与合频吸收,绝大多数生物化学样品在此区域均有相应的吸收带,因此借助这些信息可进行定性或定量分析。图2为柴胡提取液样本的原始近红外光谱。可以看出,不同样本的光谱特征并无显著差异,在6 944 cm-1和5 155 cm-1附近有两强吸收峰,分别为O—H基团的一级倍频和伸缩振动组合频区域。
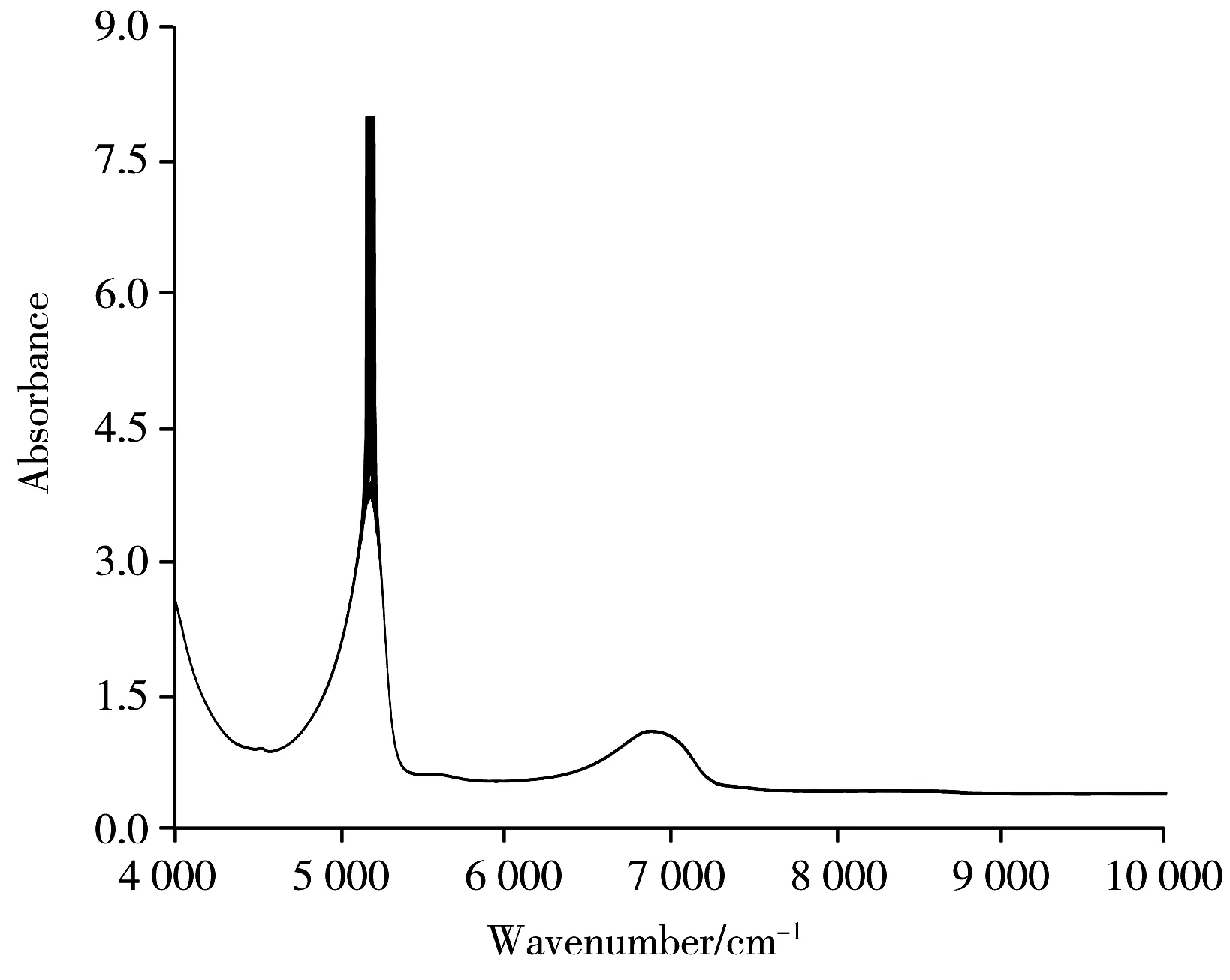
图2 柴胡提取液的原始近红外光谱Fig.2 Raw near infrared spectra of samples collected from the extract of Bupleuri Radix
2.3 定量模型的建立与评价
2.3.1 异常光谱的剔除测量环境的改变,仪器和性能参数的变化等许多不确定性因素均会导致异常光谱的产生。异常光谱的存在会影响模型的准确性和适用性,因此在建模之前,必须先排除异常光谱。本实验使用马氏距离法识别异常光谱(马氏距离是每个样本光谱与所有样本集平均光谱之间的距离,通过95%置信水平的Chauvenet检验识别样本是否异常[25])。如图3所示,126个样本中有8个样本的马氏距离超出阈值,属于光谱异常样本。异常样本的总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的含量均明显低于其余样本,4个指标平均值分别为125.92 μg/mL、5.02 mg/mL、13.40 μg/mL、2.44 μg/mL,而其余118个样本的4个指标平均值则分别为210.12 μg/mL、7.89 mg/mL、26.23 μg/mL、5.12 μg/mL。删除8个光谱异常样本后,将剩余118个样本用于后续模型的建立与验证。
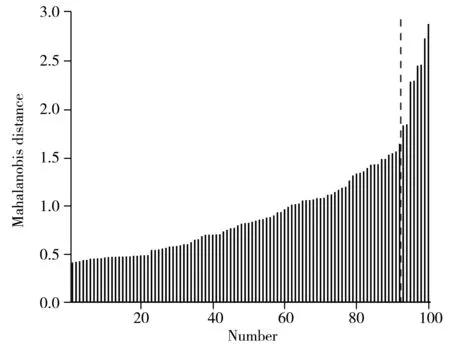
图3 异常光谱的判别Fig.3 Identification of outlier samples

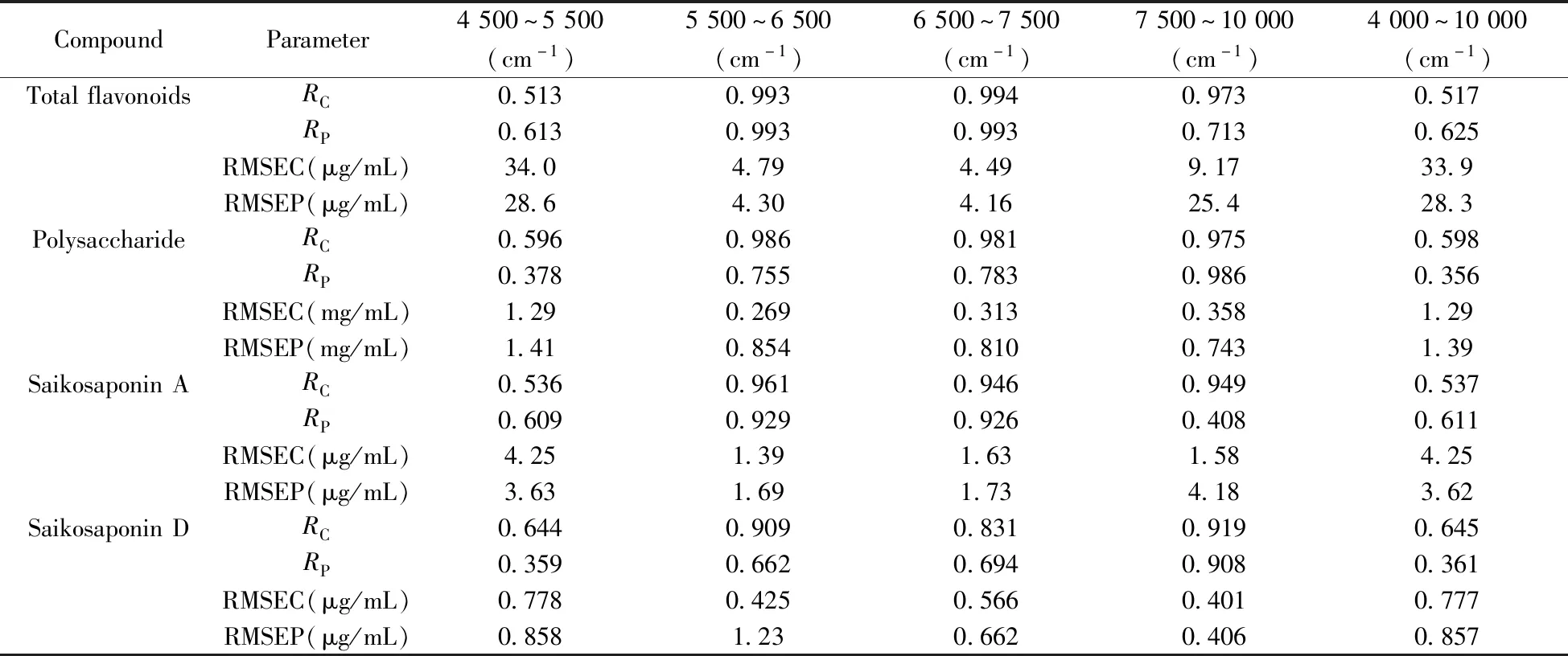
表2 近红外波段对PLS模型性能的影响Table 2 Effects of near infrared spectral section on PLS models
2.3.3 光谱预处理方法的选择基线漂移、噪声、环境、背景干扰等因素会对近红外光谱产生明显干扰[29-30],为了提高信噪比和提升定量校正模型的准确度和稳健性[31],在建模之前一般要对原始光谱进行预处理。本实验基于上述最佳光谱波段,采用无光谱数据预处理(No pretreatment)、一阶导数(1D)、一阶导数+SG平滑(1D+SG)、一阶导数+Norris平滑(1D+Norris)、二阶导数(2D)、二阶导数+SG平滑(2D+SG)、二阶导数+Norris平滑(2D+Norris)共7种不同光谱预处理方法对建模效果进行考察,结果见表3。多糖和柴胡皂苷D模型在无预处理方式下RMSEC和RMSEP值最小且相近,RP值最大,模型性能最好;总黄酮和柴胡皂苷A模型分别在选用1D+SG和2D+Norris预处理时模型效果最好。为了提升模型预测精度,总黄酮、多糖、柴胡皂苷A和柴胡皂苷D分别选择1D+SG、No pretreatment、2D+Norris和No pretreatment 4种光谱预处理方法。
2.3.4 主因子数的选择在构建PLS模型时,主因子个数的选择会显著影响模型的预测性能。主因子数过少则建模信息不全,出现“欠拟合”,导致模型精度不足;主因子数过多则会带入大量无关信息(如噪音),出现“过拟合”,导致模型预测能力明显下降[32-33]。本研究以交叉验证误差均方根(Root mean square error of cross validation,RMSECV)为评价指标,考察不同主因子数对PLS模型预测性能的影响。RMSECV值越小,代表模型预测性能越高,RMSECV最小值对应的主因子数为最佳主因子数。结果表明,总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的PLS模型分别选取主因子数为7、9、5、10时所得的RMSECV值最小。因此,本实验选择7、9、5、10分别作为总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的最佳主因子数。
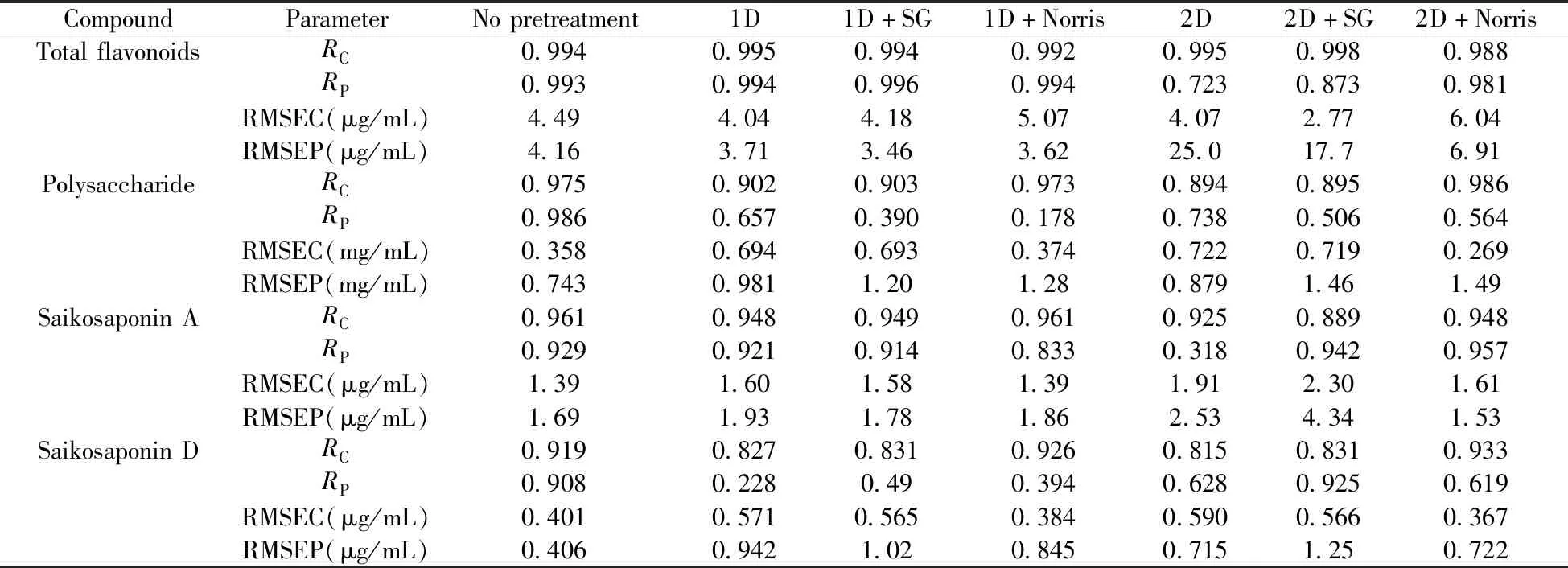
表3 不同光谱预处理方法对PLS模型性能的影响Table 3 Effects of different spectral pretreatments on models
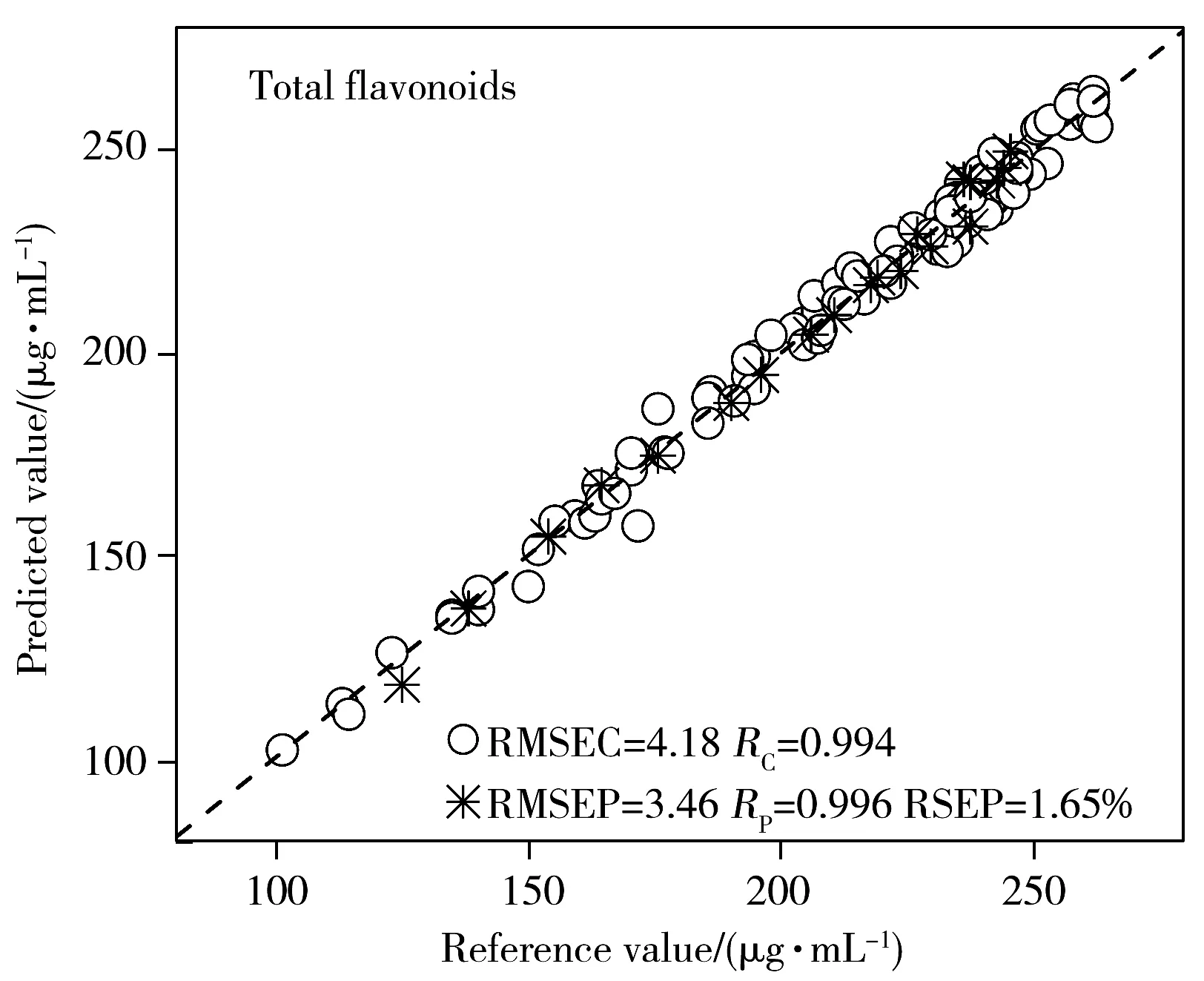
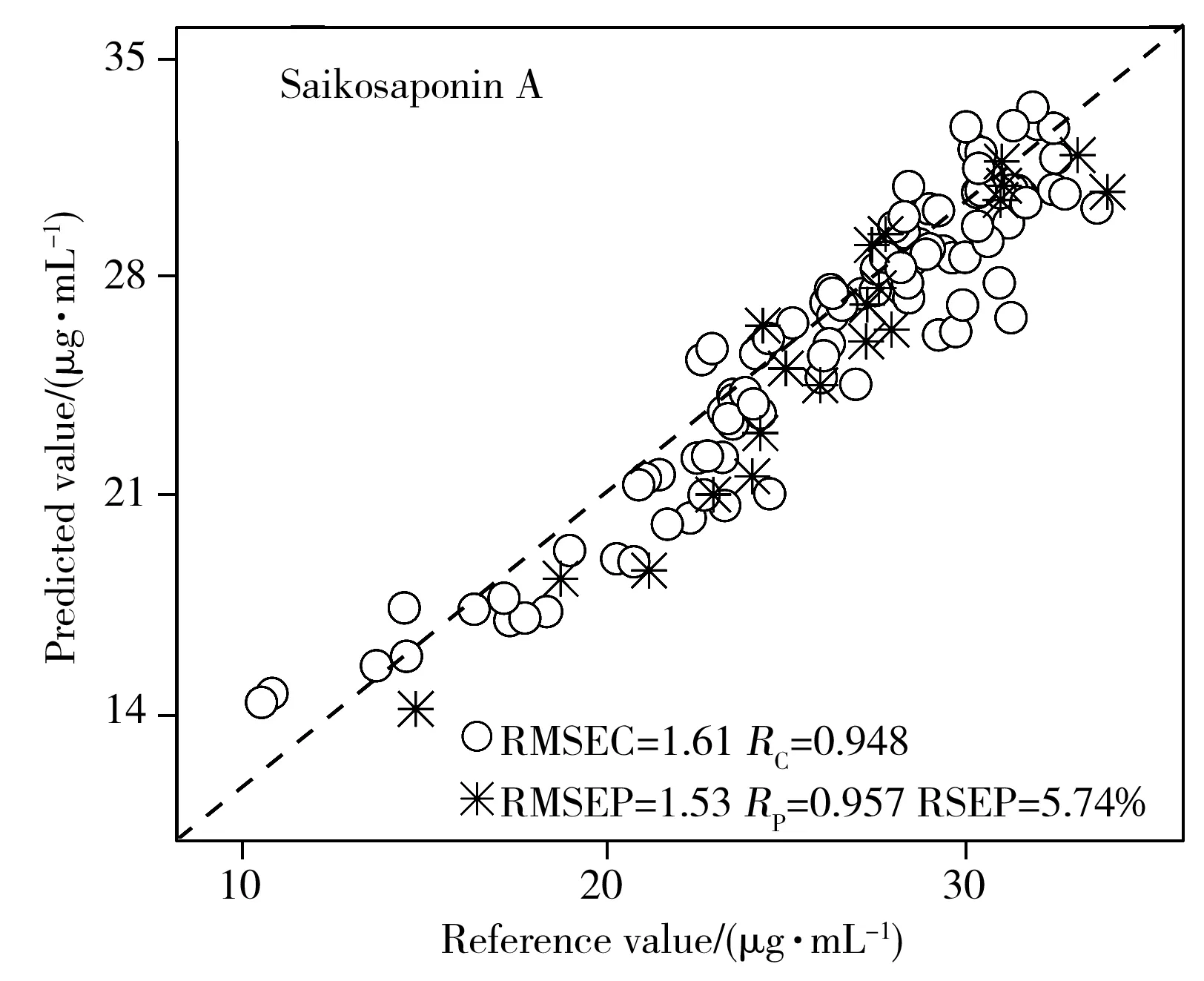
2.3.5 近红外定量模型的建立与验证基于上述优化过程,建立了总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的PLS定量校正模型。4个药效指标PLS模型的校正集和预测集中的参考值和近红外预测值的相关性见图4。从图中可以看出,近红外预测值与参考值表现出良好的拟合度,4个定量模型的RC和RP均大于0.9,RMSEC分别为4.18 μg/mL、0.358 mg/mL、1.61 μg/mL、0.401 μg/mL;RMSEP分别为3.46 μg/mL、0.743 mg/mL、1.53 μg/mL、0.406 μg/mL,每个模型的RMSEC和RMSEP较小且较为接近,并且总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的RSEP分别为1.65%、8.28%、5.74%和7.52%,均在10%以内。表明模型的预测精度较高,可准确预测柴胡提取液中总黄酮、多糖、柴胡皂苷A和柴胡皂苷D的含量。
为了进一步验证模型的预测能力,利用已校正的模型预测新的一批柴胡提取液样品。结果如图5所示,可看出模型给出的近红外预测值与参考值十分接近,变化趋势基本保持一致,表明采用NIRS可以成功预测柴胡提取过程中的4种药效指标。
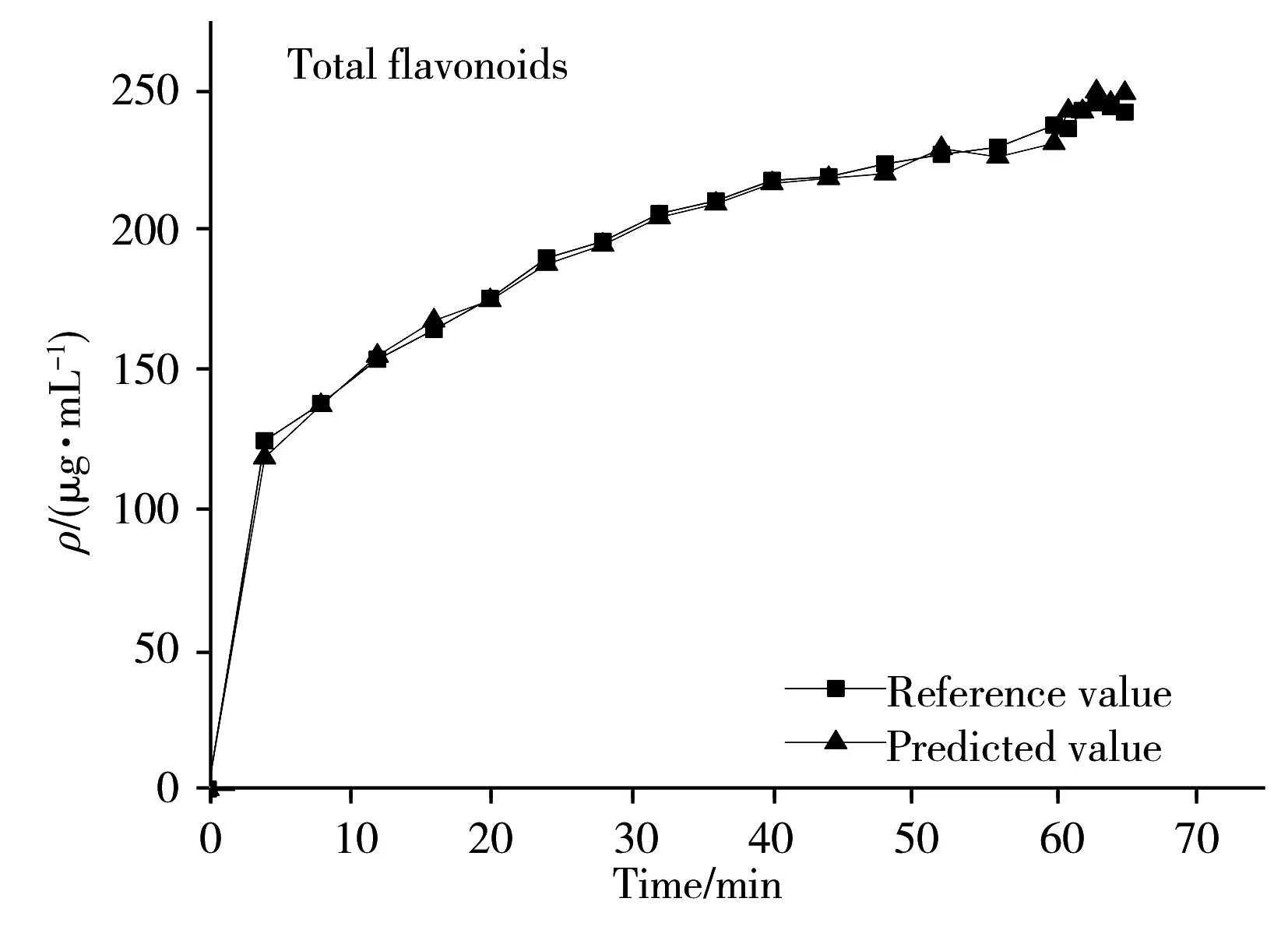
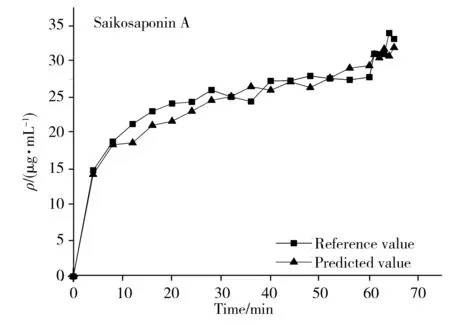
3 结 论
本文采用NIRS结合PLS法建立定量校正模型,实现了柴胡提取液中总黄酮、多糖、柴胡皂苷A和柴胡皂苷D 4种成分的快速测定。所建定量模型的R值均在0.9以上,RSEP在10%以内,模型的预测精度较高。该方法的操作简便、无需样品预处理,且成本较低,不涉及有机试剂,绿色无污染,弥补了常规分析方法的不足,可推广应用于中药提取过程的在线分析,这对于节约资源、提升中药质量控制水平具有重要的借鉴意义。
