新型杀菌剂氟唑菌酰胺合成新工艺
2020-11-04顾旻旻柴华强
尹 凯,顾旻旻,柴华强,于 江
(浙江南郊化学有限公司,浙江 绍兴 312369)
吡唑酰胺类化合物因作用机制独特、安全高效以及吡唑环上取代基变化多样而成为杀菌剂研究的热点。氟唑菌酰胺是巴斯夫公司开发的一种高选择性吡唑酰胺类杀菌剂,对病原菌具有高靶标性。其结构新颖、广谱高效,作用方式不同于现有杀菌剂,已成为杀菌剂研究的新热点[1-2]。
目前国内研究氟唑菌酰胺的合成并不多,而且基本都是以 1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯和 3',4',5'-三氟-2-氨基联苯作为合成氟唑菌酰胺的两个重要中间体。3',4',5'-三氟-2-氨基联苯的合成方法主要有以下几种:⑴ 采用3,4,5-三氟苯基溴化镁与苯基亚甲基氨基-2-氯苯为原料,经钯催化反应制得[3];⑵ 采用 3,4,5-三氟苯肼和苯胺为原料,在二氧化锰或酞菁锰催化下反应得到[4-5];⑶ 采用3,4,5-三氟苯硼酸和邻溴苯胺为原料,PdCl2(PPh3)2催化反应得到[6-7];⑷ 以3,4,5-三氟苯硼酸与邻氯硝基苯为原料,微波辅助,钯/石墨烯催化下经Suzuki偶联、氢气/雷尼镍还原得到[8]。几种合成方法如图1所示。
方法⑴中的原料苯基亚甲基氨基-2-氯苯的合成需要用到5 equiv苄醇,原子利用率低[9],催化剂制备困难,且后处理步骤繁琐,因此难以实现工业化。方法⑵反应收率偏低且需要加入过量很多的苯胺和二氧化锰,三废处理困难,此外还会产生 16%的3',4',5'-三氟-4-氨基联苯副产物,提纯困难,制备成本偏高,不符合当前绿色化学的要求;用酞菁锰为催化剂可适当提高收率,但酞菁锰价格昂贵,因此难以实现工业化。方法⑶应用的 1,4-二氧六环溶剂对环境不友好,如用甲苯替代 1,4-二氧六环,则收率降低。方法⑷反应速度快,但是需要用到3 equiv 3,4,5-三氟苯硼酸,成本高。
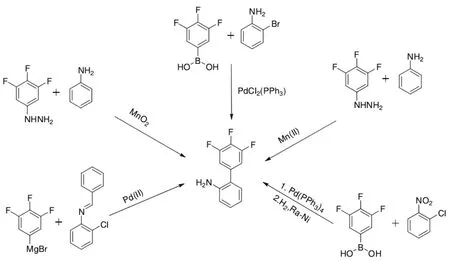
图1 氟唑菌酰胺中间体3',4',5'-三氟-2-氨基联苯的合成方法
针对上述合成方法存在的问题,本文以 1,2,3-三氯苯为起始原料,经氟化、溴化、格氏、偶联、加氢还原、缩合6步反应合成了氟唑菌酰胺(图2),总收率50%以上,含量99%以上。该工艺路线清洁、高效、可行,具有较好的工业化应用前景。
1 试验部分
1.1 仪器和试剂
仪器:Agilent 1260高效液相色谱仪;Agilent 7820A气相色谱仪。
试剂:1,2,3-三氯苯、铈改性负载型镍基催化剂[10],自制;硼酸三甲酯,山东国邦药业有限公司;其他所用试剂和溶剂均为试剂级。
1.2 试验方法
1.2.1 1,2,3-三氟苯(3)的合成
在1 L高压釜中,加入181.5 g 1,2,3-三氯苯(1.0 mol)、275 g环丁砜、208 g氟化钾 (3.6 mol)、2 g CNC+催化剂,氮气置换后,升温至170~180 ℃,压力控制在2 MPa,保温保压反应。取样合格后,精馏得1,2,3-三氟苯105.6 g,无色透明液体,纯度99%,收率80%。剩余物料继续套用,用于下一批次。
1.2.2 3,4,5-三氟溴苯(4)的合成
在 2 000 mL烧瓶中投入 132 g 1,2,3-三氟苯(1.0 mol)、255 g二氯甲烷,搅拌配成溶液,降温至10 ℃以下,加入103 g溴化钠 (1.0 mol)和120 g磷酸二氢钠 (1.0 mol)的水溶液,然后缓慢滴加10%次氯酸钠溶液890 g (1.2 mol),控制温度15 ℃以下,滴完保温反应。转化合格后,静置分层,有机层水洗至中性,脱溶得到3,4,5-三氟溴苯粗品。在-15 ℃对粗品熔融重结晶,得到200 g产物3,4,5-三氟溴苯,纯度99.5%,收率94.5%。
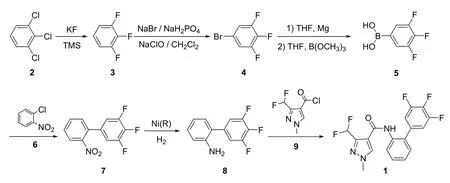
图2 氟唑菌酰胺合成新路线
1.2.3 3,4,5-三氟苯硼酸(5)的合成
在500 mL四口烧瓶中投入0.6 g镁条、30 g THF,搅拌下滴加4.2 g 3,4,5-三氟溴苯(0.02 mol),进行格氏反应的引发,生成 3,4,5-三氟苯溴化镁溶液。在上述溶液中,加入5.8 g的镁条、50 g的THF,在5~10 ℃滴加3,4,5-三氟溴苯38 g (0.18 mol),滴毕,搅拌反应2 h。再加入100 g THF,滴加43 g硼酸三甲酯 (0.41 mol),1 h内滴毕,降温至-5 ℃,保温反应2 h。然后滴入250 mL 12.5%稀盐酸,温度自然上升至25 ℃,静置分层,用150 mL甲苯萃取水层,无水硫酸钠干燥,过滤,减压蒸馏得粗品,用水洗涤后得到白色固体3,4,5-三氟苯硼酸29.1 g,纯度99.1%,收率82.6%。
1.2.4 3',4',5'-三氟-2-硝基联苯(7)的合成
在1 000 mL四口烧瓶中加入31.5 g 2-氯硝基苯(0.20 mol)、400 mL甲苯、200 mL水、45.7 g 3,4,5-三氟苯硼酸(0.26 mol)、92 g碳酸钾,氮气置换3次后,加入0.07 g二(乙酰丙酮)钯(II)。氮气保护下搅拌升温至85 ℃回流3 h。降温至室温,分层,用50 mL甲苯萃取水层3次。合并有机层,减压蒸馏得46.1 g 3',4',5'-三氟-2-硝基联苯,淡黄色固体,纯度98.0%,收率91.1%。
1.2.5 3',4',5'-三氟-2-氨基联苯(8)的合成
在1 L高压釜中投入75.9 g 3',4',5'-三氟-2-硝基联苯 (0.30 mol)、1.5 g铈改性负载型镍基催化剂和400 g甲醇。氮气置换后,通入氢气至1.0 MPa、温度80 ℃保温反应至不再吸氢,降温至室温,释放釜内压力,过滤,滤液减压蒸馏回收甲醇,残液用乙酸乙酯和水萃取后,减压蒸馏浓缩得到 65.6 g 3',4',5'-三氟-2-氨基联苯,纯度99.3%,收率98.0%。加甲苯溶解,用于下一步反应。1H NMR(400 MHz,CDCl3)δ: 6.73(d,J=8.0Hz,1H), 6.79(t,J=7.4Hz,1H),7.00-7.09(m, 3H), 7.15(t,J=7.7Hz, 1H)。
1.2.6 氟唑菌酰胺(1)的合成
在1 000 mL四口烧瓶中投入3',4',5'-三氟-2-氨基联苯(约56 g,0.25 mol)甲苯液、搅拌升温至45 ℃左右,滴加 50 g 1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯 (0.26 mol),滴完升温至70 ℃,保温反应,取样转化合格后,分别用80 g 5%盐酸、80 g 10%碳酸氢钠溶液、80 g水洗涤。搅拌下降温至室温,甲苯层析出白色固体,抽滤,烘干得产品 85.1 g。外标含量99.0%以上,收率90.5%,熔点135~137 ℃。1H NMR(400 MHz, CDCl3)δ: 3.91(s, 3H), 6.63(t, 1H),6.95-7.04(m, 2H), 7.21-7.25(m, 2H), 7.40-7.48(m,1H), 7.81(bs, 1H), 7.94(s, 1H), 8.21(d, 1H)。
2 结果与讨论
2.1 溴化反应配比的选择
此步以1,2,3-三氟苯为原料,二氯甲烷作溶剂,加入含有缓冲溶液的溴化钠水溶液,滴加次氯酸钠发生溴代反应。以 1,2,3-三氟苯与含有缓冲溶液的溴化钠水溶液摩尔投料比1∶1为反应基本条件,研究了次氯酸钠投量对溴化反应的影响,试验结果列于表1。
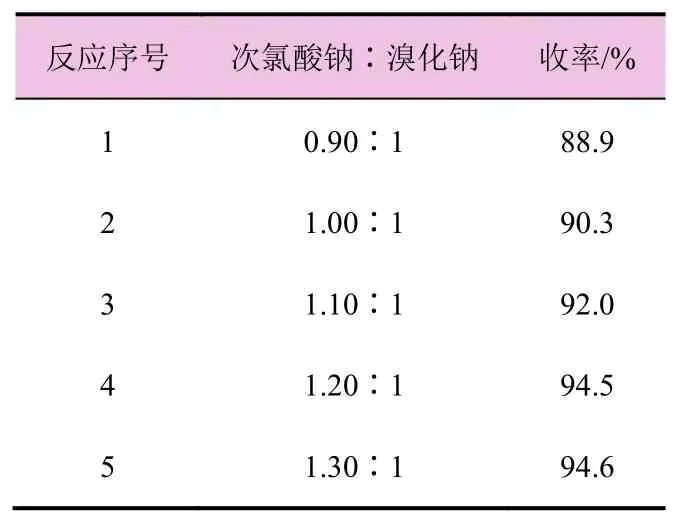
表1 次氯酸钠投量对反应的影响
由表1可以看出,随着次氯酸钠与溴化钠摩尔配比的升高,收率也随之增加,当配比达到 1.20∶1后,收率接近最高,配比再升高时,收率几乎不变。因此,选择1.20∶1为最佳反应配比。
2.2 加氢还原催化剂的优化选择
此步加氢反应条件:3',4',5'-三氟-2-硝基联苯(0.30 mol)、铈改性负载型镍基催化剂(2%,m/m)、1.0 MPa、 80 ℃。在此条件下研究了不同的催化剂对反应选择性及防脱氟效果的影响(表2)。
从表2可以看出,不同的催化剂对此反应选择性及总脱氟率的影响差异较大。笔者测试了Ni(R)、Ni(R)+乙醇胺、Ni(R)+双氰胺及铈改性负载型镍基催化剂4种常用的催化体系,效果相差较大,其中以铈改性负载型镍基催化剂效果最佳,所以选择铈改性负载型镍基催化剂作为此步加氢还原反应的催化剂。
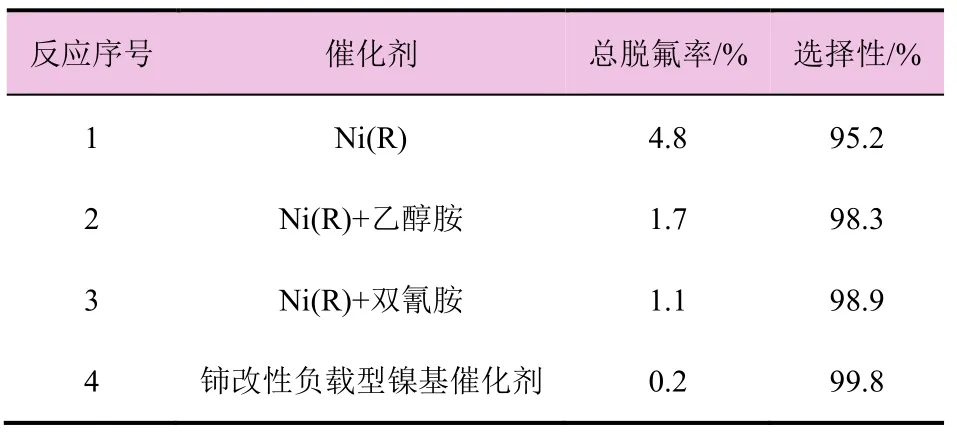
表2 不同催化剂对选择性及防脱氟效果的影响
2.3 缩合反应条件的优化选择
此步骤是以中间体 3',4',5'-三氟-2-氨基联苯和1-甲基-3-二氟甲基-1H-吡唑-4-甲酰氯为原料,在甲苯做溶剂的条件下进行缩合反应,研究了在两种原料配比一定的情况下,反应温度对反应时间、含量及收率的影响(表3)。
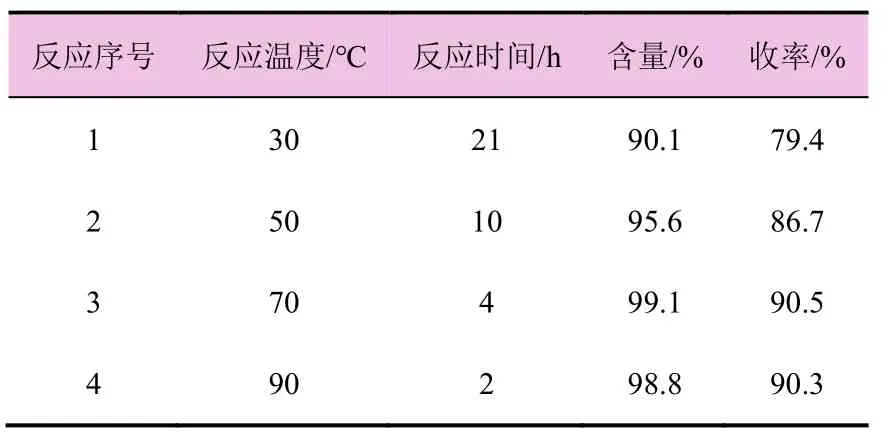
表3 反应温度对缩合反应的影响
从表3可以看出,不同的反应温度对反应时间、含量和收率的影响较大。温度太低,转化不完全,导致含量和收率偏低;温度过高,反应时间缩短,但副反应增多,导致含量和收率开始下降。最终确定了缩合反应的最佳条件为反应温度70 ℃,反应时间4 h。在此条件下,缩合反应的含量和收率均达到最佳。
3 结 论
本合成方法以1,2,3-三氯苯为起始原料,经氟化、溴化、格氏、偶联、加氢还原、缩合6步反应合成了氟唑菌酰胺,总收率50%以上,含量99%以上。本法与其他文献报道的方法相比具有如下优点:⑴ 操作简便,原料廉价易得;⑵ 总收率高,产品纯度高,杂质少;⑶ 用铈改性负载型镍基催化剂替代常规的催化剂,提高了反应选择性,大幅降低了总脱氟率;⑷ 用1,2,3-三氯苯合成3,4,5-三氟溴苯,大大缩短了合成步骤,同时也避免了硝化、重氮化等危险工艺,减轻三废压力同时也更安全稳定。
