Colonization of neonate mouse spermatogonial stem cells co-culture with Sertoli cells in the presence and absence soft agar
2020-11-03AliTalebiShadanNavidMaryamBorhaniHaghighiYumiHoshinoMehdiAbbasiZahraKhosravizadeh
Ali Talebi, Shadan Navid, Maryam Borhani-Haghighi, Yumi Hoshino, Mehdi Abbasi, Zahra Khosravizadeh
1School of Medicine, Shahroud University of Medical Sciences, Shahroud, Iran
2Tissue Engineering and Stem Cells Research Center, Shahroud University of Medical Sciences, Shahroud, Iran
3Sexual Health and Fertility Research Center, Shahroud University of Medical Sciences, Shahroud, Iran
4Department of Anatomy, Gonabad University of Medical Sciences, Gonabad, Iran
5Department of Anatomy, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
6Laboratory of Reproductive Endocrinology, Graduate School of Biosphere Science, Hiroshima University, Hiroshima, Japan
ABSTRACT
KEYWORDS: Spermatogonial stem cell; Soft agar; Colonization;Proliferation; Mouse
1. Introduction
Spermatogonial stem cells (SSCs) simultaneously expand and differentiate into other cells, leading to sperm production. These cells are considered as the foundation for sperm production and male fertility. However, like tissue-specific stem cells, the number of these stem cells is very low[1]. Many reports have indicated a high percentage of male infertility, particularly in children, after cancer treatment by chemotherapy or radiotherapy[2]. In vitro proliferation of SSCs has paved way for investigations into the treatment of male infertility. Sertoli cells play a vital role in the regulation of proliferation and differentiation of SSCs. The presence of Sertoli cells increased the survival rate of SSCs in cultured mouse testis tissue and cell samples[3]. Previous studies investigating the factors associated with the proliferation of cultured SSCs have focused on growth factors such as leukemia inhibitory factor[4], glia cell line-derived neurotrophic factor[5], basic fibroblast growth factor,epidermal growth factor[5,6], and stem cell factor[7,8]. Many studies have confirmed that these factors cause the development of SSCs when applied in different culture systems. One of the most widely used techniques for in vivo cellular transformation is the soft agar colony formation assay. Other techniques such as the clonogenic assay have also been used by researchers for evaluating the ability of cells to form colonies[9]. Because the soft agar culture provides a suitable environment and similar in vivo for cell proliferation and migration. In recent years, this culture medium has been known to identify tumorigenic inhibitors in cancer cells[10].
Previous studies have reported clonal proliferation of bone marrow cells and lymphocytes in soft agar culture and the factors involved in the regulation of proliferation and differentiation in these cells[11,12];however, not much research has been done in the effects of soft agar on the in vitro proliferation of SSCs in two-dimensional culture.
This study was to investigate the in-vitro effects of soft agar supplemented with leukemia inhibitory factor and glia cell linederived neurotrophic factor on the proliferation of neonatal mice SSCs co-cultured with Sertoli cells in two-dimensional culture.
2. Methods and materials
2.1. Animals
SCCs were harvested from 3 to 6-day old NMRI male mice. The mice were kept under standard conditions with free access to food and water. We kept mice at 12 h in the dark and 12 h in the light and at constant temperature and humidity.
2.2. Isolation of neonatal testis cells
Testis cells obtained from 3 to 6-day old NMRI male mice after general anesthesia had been induced with chloroform (Sigma St.Louis, USA, C2432). Testicular cells were isolated by two-step enzymatic digestion (5 mice at a time in each group). Testes were removed from the scrotum and transferred to phosphate buffered saline (PBS) (Sigma, Munich, Germany). The testes were cut into small pieces after the removal of tunica albuginea. The minced testicular tissues were transferred to a digestion solution containing collagenase type Ⅳ(1 mg/mL, Sigma, Steinheim, Germany),deoxyribonuclease (10 μg/mL, Sigma, St. Louis, USA), and hyaluronidase (0.5 mg/mL, Sigma, St. Louis, USA) for 20 min at 37 ℃ in a 5% CO2incubator. To disperse the tubule cells, pipetting was performed at every 2-5 min until the tubules were separated.The dispersed tubule cells were centrifuged for 5 min at 1 500 × g.The same procedure and enzymes were used for the second step of the enzymatic digestion (15 min). The separated cells were washed with PBS (Sigma)[4,13,14].
2.3. Flow cytometry
After the two-step enzymatic digestion, a single-cell suspension from the samples was obtained. These cells were predominantly SSCs. The cell suspension was centrifuged at 1 500 × g for 5 min after which the cells were counted. A density of 2×105cells/cm2was transferred into a petri dish and kept for 24 h at 37 ℃ in a 5% CO2incubator. Following incubation, the dish was washed and the cells were collected.
Flow cytometry (Thermo Fisher Scientific, US) was performed by using promyelocytic leukaemia zinc finger protein (PLZF)antibody to evaluate the purity of the SSCs. To identify PLZF positive cells, 10 μL primary antibody (Anti-PLZF antibody;Abcam Inc., Cambridge, MA, USA, rabbit polyclonal to PLZF)was added to the cells and incubated for 1 h at room temperature.After permeabilization with 0.4% Triton ×100 (Sigma) and washing in PBS, 10 μL of secondary antibody (Donkey Anti-Rabbit;Abcam Inc., Cambridge, MA, USA) conjugated with fluorescein isothiocyanate was added (1 h, 4 ℃). Cells used as control were not treated with any antibody[15,16].
2.4. Preparation of soft agar-coated dishes
The soft agar coated dishes were prepared as follows: 0.7%(w/v) agar (Fisher Scientific, Loughborough, United Kingdom)was dissolved in distilled water to form a gel. Subsequently, the solution was mixed with the same volume of the basic culture medium (Sigma, St. Louis, USA) to achieve a final concentration of 0.35%. Then, 30 mm dishes were coated with 1 mL of the final soft agar (0.35%) solution and stored in an incubator at 37 ℃ at 5% for 1 h[4,17].
2.5. SSCs culture
SSCs were cultured for two weeks on a 24-well plate (density of 2×105cells/cm2) in a basic culture medium- minimum essential mediumα(sigma St. Louis, USA) containing 10% fetal bovine serum(Sigma, Deisenhofen, Germany), 1× nonessential amino acids(Invitrogen, Carlsbad, CA, USA), 0.1 mM 2-mercaptoethanol(Sigma, Deisenhofen, Germany), 100 U/mL penicillin, and 100 μg/mL streptomycin (both from Sigma, Deisenhofen, Germany), along with 103U/mL human recombinant leukemia inhibitory factor (B&D,Franklin Lakes, NJ, USA) and 10 μg/mL glial cell line-derived neurotrophic factor (R&D, Emeryville, CA, USA) supplementation in each culture dish. In the experimental group, soft agar-coated dishes were used for the co-culture of SSCs and Sertoli cells,but in the control group, soft agar was omitted. Culture medium replacement was performed every 2 days. Culture dishes were kept in an atmosphere humidified with 5% CO2at 32 ℃. Cells were cultured for 7 days after which colony staining was performed by using alkaline phosphatase[3].
2.6. Alkaline phosphatase staining
For assessing alkaline phosphatase activity, the cells were stained by using Fast Red TR/Naphthol AS-MX Tablets (Sigma St. Louis,USA, F4648), according to the manufacturer’s instructions. Alkaline dye was prepared by briefly dissolving a Fast Red TR/Naphthol ASMX Tablets in 1 mL deionized water. The alkaline dye was added to the cells and incubated at 37 ℃ for 30 min. The stained cells were observed under an inverted microscope (BX51, Olympus, Japan) at×100 magnification following incubation.
2.7. Immunocytochemistry staining for characterization of SSCs colonies
SSCs colonies were fixed in 4% paraformaldehyde for 20 min,and were permeabilized with 0.4% Triton X100 (Sigma, Germany)and blocked with 10% goat serum (Sigma, Germany). Primary antibody was added to the cells and incubated for 2 h at 37 ℃ with rabbit polyclonal Anti-PLZF antibody (dilution 1:100; Sigma,Germany). Subsequently, the cells were washed with PBS, and the secondary antibody, Donkey Anti-Rabbit labeled with fluorescent isothiocyanate diluted at 1:100 (Sigma, Germany), was added for 3 h. Control cells were not treated with the primary antibody. Nuclei staining was performed by using 4,6-diamidino-2-phenylindole(1 μg/mL; Sigma, Germany).
2.8. Colony assay
The diameter and number of colonies were determined at the end of the second week (on day 14 of culture). After the cultivation of total spermatogonial cells in the 30 mm dishes, colonies were formed on the 4-5th day in the experimental group and on the 7th day in the control group. All colonies in each 30 mm dish were counted for each group. All measurements were repeated three times. We measured the diameter of the colonies by using ocular grid on the inverted microscope used to observe the images. The images were analyzed by using ImageJ software, version 1.6 (National Institutes of Health, Bethesda, MD, USA).
2.9. Real-time PCR
After culturing the cells for 2 weeks, we used the real-time polymerase chain reaction (PCR) technique to evaluate the expression of promyelocytic leukaemia zinc finger protein (PLZF) in the undifferentiated cells, and DNA-binding protein inhibitor (ID-4)and tyrosine-protein kinase kit (c-kit) in the differentiated cells.Following the manufacturer’s protocol, the Trizol reagent (Ready Mini Kit, Qiagen, USA) was used for total RNA extraction.Real-time PCR was performed by using 1 μg of total RNA. The cDNA synthesis kit (Transcript First Strand cDNA Synt, Roche,Indianapolis, IN, USA) was used for this purpose, according to the manufacturer’s protocol. Forty PCR amplification cycles were performed for the samples. The results were analyzed by using Applied Bioscience 7500 Fast with SYBR Green detection.Nonspecific PCR products and primer dimers were detected by performing melt curve analysis after each PCR run. The comparative CT method (ΔΔCt) was used for normalizing the samples against glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used an internal control. The primer sequences were been shown in Table 1[18].
2.10. Statistical analysis
Results were expressed as mean±standard deviation (mean±SD).Statistical analysis was performed by using unpaired t-test for the comparison of the diameter and number of colonies and gene expression studies in the experimental and control groups. P<0.05 was considered statistically significant.
2.11. Ethics statement
This study was approved by Ethical Committee of Shahroud University of Medical Sciences on 4 March, 2020 (No. IR.SHMU.REC.1398.166). Moreover, handling of animals was carried out in accordance with the guidelines of the Iranian Council for Use and Care of Animals.
3. Results
3.1. Purification of SSCs
To determine the purity of SSCs, we performed flow cytometry by using PLZF antibody after the enzymatic digestion. Of all cells,72.7% cells expressed PLZF (Figure 1). The other testicular cells isolated from the testes were transferred and co-cultured with the SSCs. The majority of these cells were Sertoli cells which created feeder monolayer cells, and played a vital role in the formation of colonies of SSCs. Following proliferation, SSCs formed prominent
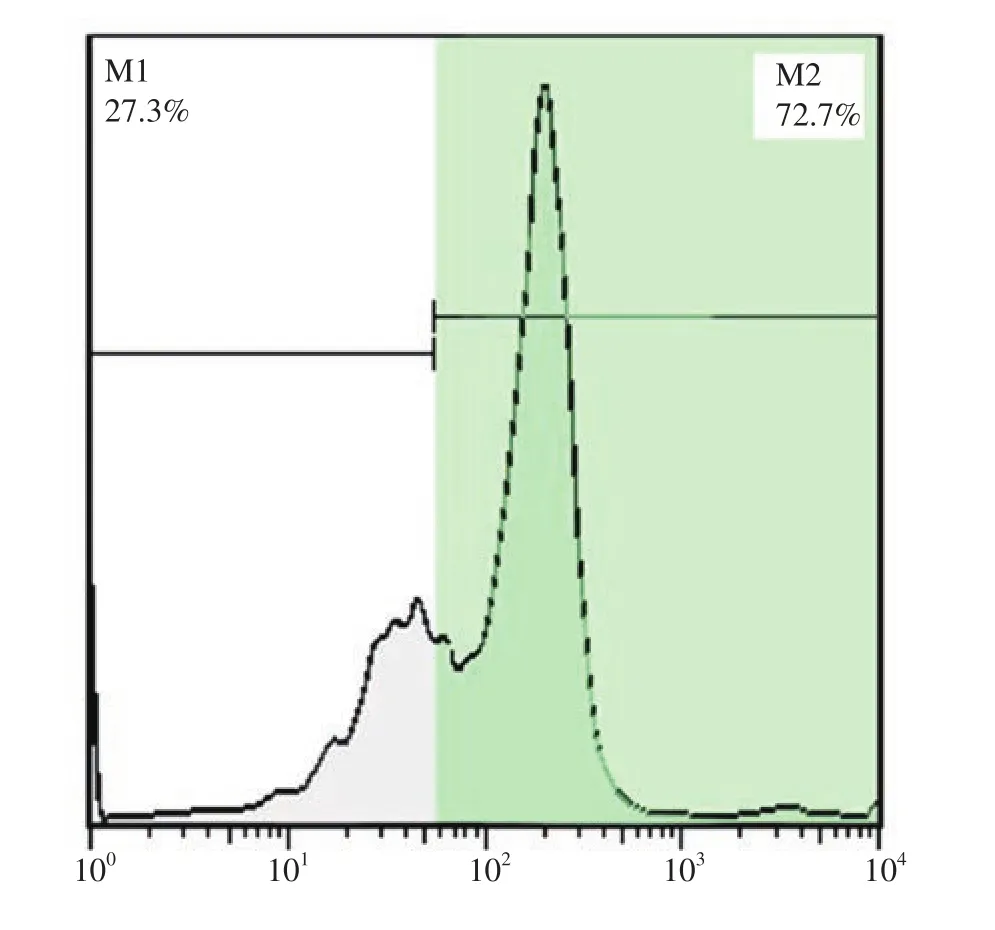
Figure 1. Flow cytometry analysis for the detection of percentage of spermatogonial stem cells purity with promyelocytic leukaemia zinc finger protein (PLZF) marker. M1: without PLZF antibody; M2: with PLZF antibody.
3.2. SSCs culture
The single-cell suspension was cultured for two weeks following the enzymatic digestion. After day 1 of culture, no colonies formed.The cells remained single and attached to the bottom of the dish.Colonies appeared on the 4-5th day of culture. Colony parameters(number and diameter) were measured at the end of the second week(the 14th day after seeding) in the two groups (Figure 2 A, B). SSC colonies formed on the 4-5th day in the experimental group and on the 7th day in the control group. These colonies were positive for the activity of the embryonic stem cell marker, alkaline phosphatase(Figure 2C).
Moreover, the identity of the colonies of SSCs was confirmed by the detection of PLZF protein expression in the nuclei of the SSCs by immunocytochemistry technique. The presence of this protein in the nucleus of clonal cells proved that these colonies were in the stage of proliferation and self-regeneration (Figure 3).
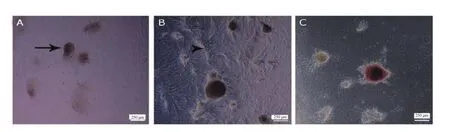
Figure 2. Inverted microscopic morphology of spermatogonial stem cells (SSCs) obtained from 3-6 day old male mice (five mice at a time in each group)(magnification ×100). The size of the colonies at the end of the second week is shown. In the control group (A) and the experimental group (B), Sertoli cells proliferate and create a monolayer of cells as a feeder layer (arrow tip), while SSCs create a prominent colony (arrow) on top of the feeder layer in both groups, with the difference that the experimental group contains soft agar. After cultivation of SSCs, diameter and number of colonies increase in both groups particularly in the experimental group due to the soft agar and they are positive for alkaline phosphatase activity (C).
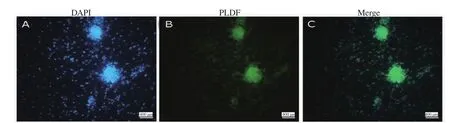
Figure 3. Immunofluorescent staining of colonies of spermatogonial stem cells (SSCs) (magnification ×100). Nuclei are stained with 4,6-diamidino-2-phenylindole (DAPI) (blue color) (A); SSCs are positive for promyelocytic leukaemia zinc finger protein (PLZF) in the nuclei (green color) (B), and a merge of A and B in C.
3.3. Results of colony assay
From the colony assay results, the average number of colonies was 4.4±1.9 and the diameter was (257.3±9.1) μm in the control group.In the experimental group, the number and diameter of colonies were 6.5±2.1 and (339.7±15.4) μm respectively at the end of the second week. The number and diameter of the colonies formed in the experimental group were significantly increased as compared with those of the control group (P<0.05, P<0.01, respectively) (Figure 4).
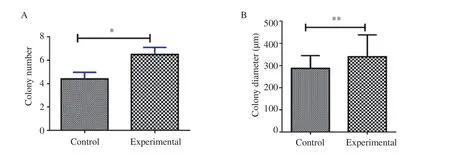
Figure 4. Comparison of colony number (A) and diameters (B) between the control and experimental groups in the second week. *P<0.05, **P<0.01.
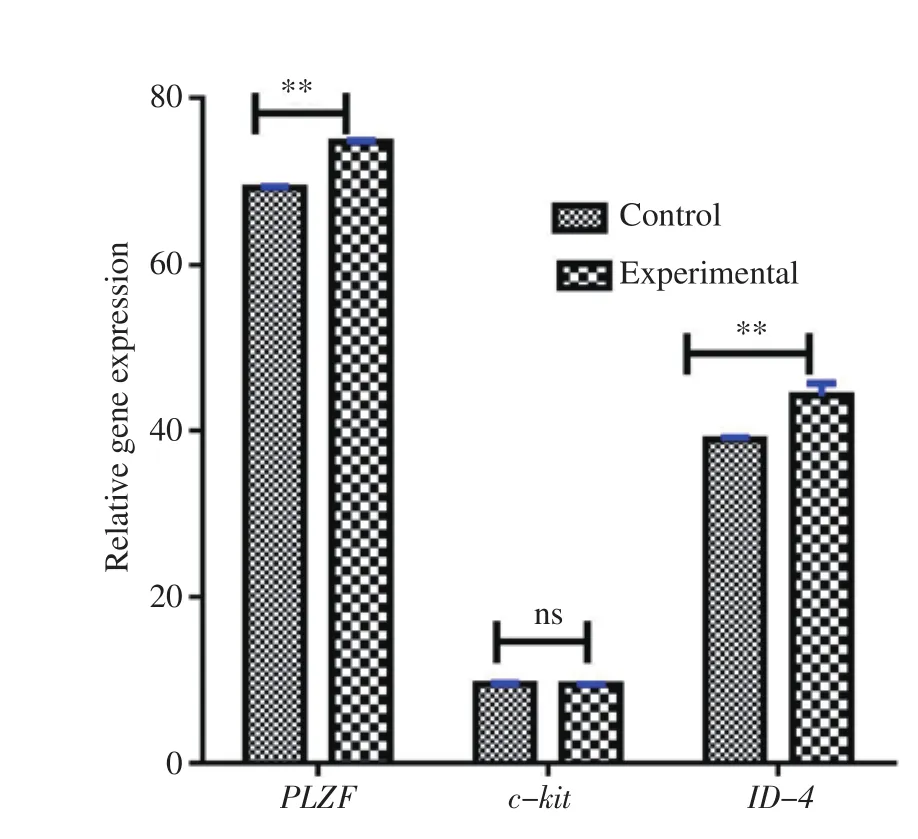
Figure 5. Expression pattern of c-kit, PLZF, and ID-4 genes after two weeks of culture analyzed by real-time PCR. Levels of ID-4 and PLZF in the experimental group have an increased value as compared with the control group, but the level of c-kit in the experimental group has no significant difference as compared with the control group. Y-axis shows ΔΔCt values of genes. **P<0.01. ns:no significant difference.
3.4. Gene expression
Expression levels of PLZF, ID-4, and c-kit genes in the SSCs cocultured with Sertoli cells were studied by using real-time PCR. The expression levels of PLZF and ID-4 genes were significantly higher in the experimental group when compared with the control group(P<0.01). The level of expression of c-kit gene was lower in the experimental group as compared with the control group, however,the difference was not significant (P>0.05) (Figure 5).
4. Discussion
In this study, soft agar coated dishes were used to investigate SSCs co-culture with Sertoli cells.Sertoli cells secrete glia cell line-derived neurotrophic factor which plays a critical role in the regulation of the self-renewing and survival of SSCs. Leukemia inhibitory factor also has a pivotal role in the regulation of the function of Sertoli cells and SSCs. Previous studies have demonstrated that the presence of leukemia inhibitory factor in a culture medium inhibits the differentiation of embryonic stem cells[19] and SSCs[3]. The effects of these growth factors on these cells have been studied by many researchers[14,20,21].
In previous studies, Sertoli cells have been utilized as a feeder layer consisting of a row of monolayer cells on the bottom of the dish on which SSCs are colonized[14,21-23]. This finding is in agreement with our results in which the presence of soft agar in the culture dishes helped to keep the feeder layer, thereby contributing to the growth and proliferation of SSCs.
Colony assay has been used as an in vitro morphological criterion to evaluate the development of SSCs in many studies[14,23,24].According to our results, the number and diameter of SSCs colonies increased at the end of the second week of culture. Razi et al also reported an increase in the number and diameter of colonies of SSCs when stem cells were co-cultured with Sertoli cells in collagencoated dishes in the presence of growth factors[21]. In other studies,colony evaluation has been used as a criterion in the proliferation and self-regeneration of SSCs. In these studies, evaluation of SSC colonies has been evaluated[14,24].
In the soft agar colony formation assay, cell clusters were gently dispersed by pipetting into a single cell suspension and were cultured in the presence of Sertoli cells acting as a feeder layer or in a medium that contains growth factors. Since the formation of colony of tumors in a two-dimensional culture medium is very slow and difficult, the soft agar colony formation assay is one of the most suitable techniques for colony production[10,26].
In recent years, three-dimensional soft agar culture system has been suggested as an efficient method for the proliferation of SSCs in vitro. This culture system is composed of two layers: soft layer and solid layers. The main disadvantage of this method is the inability to isolate live cells[4,17,25,26].
Apart from the studies of Razi et al and Barralet et al, there is a general lack of research on the appropriate dish content which is a valuable source for the development of SSCs[21,27]. In this study, we used PLZF antibody to evaluate the purity of SSCs, and we achieved a purity of 72.7%. Many studies have demonstrated the importance of PLZF antibody in the purification and identification SSCs colonies[23,28]. Our results show that soft agar has a positive effect on the expression of ID-4 and PLZF genes, and a negative effect on the expression of c-kit gene. The results of this study indicate a positive correlation between the proliferation of SSCs and culture in soft agar medium.
C-kit gene is expressed in the early stages of meiosis, so decreased expression of this gene confirms increased proliferation of SSCs.This key gene has been studied in many studies for lack of differentiation in these cells[29]. PLZF is expressed in the SSCs of Aal, As, Apand plays an essential role in regulating the proliferation of SSCs[3]. Therefore, the use of PLZF in both real-time PCR technique for gene analysis and immunocytochemistry staining for protein expression proves that these SSC colonies did not enter the differentiation stage. We also used alkaline phosphatase staining, a common technique for proving the presence of colonies.Other studies have used different genes such as Stra8 to study the formation of SSC colonies[14].
ID-4 is one of the genes that is expressed in the early stages of proliferation of most stem cells. In most studies, it is evaluated as a key gene in stem cells[30]. There are many reports that are consistent with the results of our research[4,14,21,23,31].
In recent years, the soft agar in the three-dimensional model has been mentioned to improve the culture and differentiation of SSCs.But, there is no access to living cells in the three-dimensional model of soft agar[4,32,33]. This shows the superiority of our research.
Spermatogenesis in men begins at puberty, therefore, chemotherapy and radiotherapy in children with cancers can cause loss of spermatogonial stem cells, leading to infertility. So, improving SSCs culture can provide a valuable tool for preserving fertility before cancer treatment. In recent years , stem cells, including SSCs[34] have been used to treat many diseases[16,18,35-37]. Culturing techniques provide a suitable method for the proliferation of stem cells.
Even though culturing stem cells have a number of advantages, the method has few drawbacks. There is a possibility of contamination due to the use of soft agar in the culture medium such. Also, the separation of colonies from the soft agar can a daunting task.
In conclusion, our findings indicate that SSCs obtained from neonatal mouse testis can be successfully cultured in soft agar-coated dishes. The culture protocol used in this study can increase the expansion of SSCs and can be a valuable method for future studies.
Conflict of interest statement
The authors declare no conflict of interest in any form.
Acknowledgments
We are grateful to Shahroud University of Medical Sciences for financial support of this project.
Funding
This study was supported by Shahroud University of Medical Sciences (Grant No. 98131).
Authors’ contributions
Shadan Navid conceived of the study, performed animal treatments,collected and analyzed specimen and analyzed/interpreted data.Mehdi Abbasi, Shadan Navid and Ali Talebi analyzed real time PCR.All authors conceived of and oversaw the study, interpreted data,assisted with figure preparation, and wrote the manuscript. Yumi Hoshino, Maryam Borhani and Shadan Navid read and approved the final manuscript.
杂志排行

Asian Pacific Journal of Reproduction的其它文章
- Pregnancy-associated glycoproteins as a potential marker for diagnosis of early pregnancy in goats: A scoping reviewing
- Time-lapse videography reveals different morphokinetic profiles of human embryos displaying direct or reverse cleavage at different stages of development: A retrospective sibling embryo study
- The leaf extracts of Camellia sinensis (green tea) ameliorate sodium fluoride-induced oxidative stress and testicular dysfunction in rats
- Overexpression of tyrosine phosphorylated proteins in reproductive tissues of polycystic ovary syndrome rats induced by letrozole
- Hepatic and reproductive toxicity of sub-chronic exposure to dichlorvos and lead acetate on male Wistar rats
- Vitamin D3 supplementation influences ovarian histomorphometry and follicular development in prepubertal albino rats