基于遗传算法与支持向量机的癌症特征基因提取
2020-11-02唐铭一郑虹韩立权
唐铭一 郑虹 韩立权

摘要:针对癌症基因特征提取问题,根据遗传算法中不同迭代时期的种群特性,设计了新的突变方法。多突变基因库与种群代数相关的设计,使得算法能够较快地收敛到最优解而又避免其过早陷入局部最优解中;选择算子中包括个体对种群的基因丰富度贡献;针对种群中大量的重复个体,加入重复控制,去除重复个体,提高个体与种群基因的多样性。算法在几种实验数据集上均取得了较好的结果。
关键词: 遗传算法; 支持向量机; 特征提取; 选择算子; 变异算子
中图分类号:TP181 文献标识码:A
文章编号:1009-3044(2020)26-0010-03
Abstract: A new mutation method was designed according to the population characteristics of different iteration periods in genetic algorithm to solve the problem of cancer gene extraction. Multi-mutation gene bank is designed to related with population algebra, so the algorithm converge to the optimal solution quickly and avoid falling into the local optimal solution too early. Selection operator is designed to relate with population, including the contribution of individuals to the genetic richness of the population, the MIC evaluation of individuals, and the redundancy of genes within individuals, which makes the algorithm pay attention to both the population and the characteristics of individuals. The genetic diversity of individuals and populations are improved by eliminating duplicates.
Key words:genetic algorithm; support vector machine; feature extraction; selection operator; mutation operator
1 引言
基因芯片,又称DNA微阵列,是利用核苷酸杂交技术检测生物基因的表达,采用高度集成的方法,将事先设计好的核苷酸序列组合成微阵列,以达到高通量检测的目标,是一个融合生命科学、计算机科学、化学等多学科高度结合的技术,广泛应用于基因测序、生物表达分析、癌症致病基因的发现与分析等方面[1]。利用基因芯片技术进行癌症分类,不仅可以帮助诊断疾病,而且可以帮助研究者了解疾病在分子层面上的成因。但由于微阵列数据的特殊性,使得基因芯片数据集具有少样本,高维度的特点,过高的维数会产生维数灾难的问题[2],而且数量众多的特征里又存在很多冗余和噪声基因,使得分类器不能以较高的准确率分类,同时基因芯片数据集通常是多患病样本,这造成分类样本不平衡的问题[3-4],分类器一般基于数据是平衡的这一假设,这些也是大部分研究者正在着手解决的问题。
对于基因芯片数据集样本少、高维度、高冗余的特点,一般要对数据进行特征提取,根据特征提取方法与分类器结合方式的不同,又分为filter和wapper方法[5],其中filter方法的特征提取与分类器分离,仅仅利用数据的数字特征进行相关性分析,剔除无效特征,这种方法与分类器无关,扩展性强,并且轻量,但筛选出的特征基因在不同分类器上的分类效果差異性大,筛选出的特征的分类效果一般。wapper方法,分类器与特征提取算法是一体的,特征评分标准是分类器的分类效果,因此提取出的特征,对特定分类器的分类效果是最优的。
目前比较常用的特征提取算法有遗传算法与支持向量机(svm)结合[6-7],粒子群算法与svm结合,退火算法与svm结合的,等等。本文根据遗传算法中不同迭代时期的种群特性,设计了新的突变方法,在选择算子中加入种群相关的评价方法,结合svm给出了一种特征基因提取算法。
2 特征基因提取的遗传算法设计
2.1 染色体编码
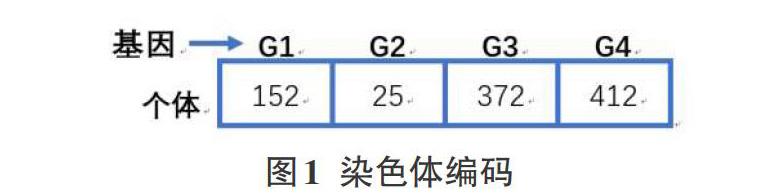
遗传算法的第一步是要构建染色体,代表问题解集中的一个解,将染色体的长度设置为所提取的特征子集的大小,每个位置的数值代表基因的索引,如图1所示。
2.2 种群的初始化和适应度函数
本算法对种群进行随机初始化,即每个染色体的基因都是随机生成的,并且染色体中没有重复的基因。种群大小设置为100,适应度函数使用svm分类器的分类准确度给出,进行5折交叉验证,即首先将数据集5等分,测试集依次取5等分的一份,生成5种不同的训练和测试集[8],然后同时训练5个分类器,并对5个分类器在测试集上的分类准确度加和求平均得到个体的适应度。
2.3 遗传算子设计
2.3.1 选择算子
选择适应度排名前10%的个体为精英个体,精英个体全部保留为下一代个体。
2.3.2 交叉算子
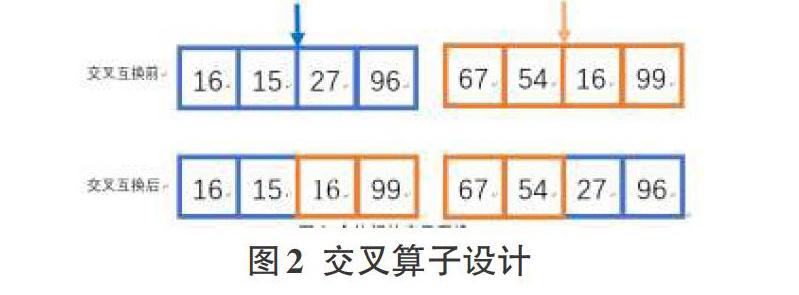
采用中点交叉互换法,如图2所示,从种群中随机选出两个个体,以解的中点为界,交叉互换两个解的一部分。本算法中交叉互换率设置为0.7,本算法在此步添加了重复控制,当chromosome中包含重复基因时,此次交叉互换失效,重新进行。
2.3.3 变异算子
变异过程是增加种群基因丰富度的关键步骤,本算法在这里进行了仔细的设计。
采用单基因突变,如图3所示,随机选取解的一个基因进行突变,本算法在突变的过程中添加了重复控制,突变时进行检查,如果突变基因在原来的解中,则需要重新进行突变,这一步和交叉互换中的重复控制保证了所有的染色体符合解约束(特征子集数),突变率设置为0.3。
本算法设计了一个基于最大信息系数(MIC)打分,并与种群代数相关的突变方法进行突变基因的选择。MIC又称最大相互信息系数,是基于信息的非参数性方法,用于衡量两个变量之间的线性或非线性的强度。本算法先利用MIC计算基因与类别的相关性,并将相关性作为基因的分数。
突变基因的集合称为突变基因库,算法设计了两个突变基因库,每次突变只以一定概率选择其中的一个基因库,同时选择概率也随种群代数变化而变化。
1)优势基因库,挑选MIC打分排名前25%的基因构成此库,并且打分越高的基因其在库中的重复基因就越多。具体是这样设计的:
上式中G为基因库,[gi]为第i个基因,基因库由MIC分数前25%的基因组成,[gscorei]为第i个基因的MIC分数,[Sumscore]为[G]中所有基因的分数和,[ gweighti]为基因[gi]的权重,[gweightmin]为G中最小的权重,[Gsize]为基因库的大小(包含重复基因),其中满足[gscorei=gscoremin]的基因[gi]不重复,[gnumi]为G中基因[gi]重复的次数,优势基因库由[gi]和[gnumi]确定。
2)全量基因库,由所有基因构成,每个基因等概率被选择。
选择突变基因库的策略是随着种群代数越多越倾向于选择全量基因库,这是因为优势基因库的作用是向种群中快速引入优势基因,同时使得算法的随机性减小,后期倾向于以大的随机性选择突变基因,目的是防止陷入局部最优点,选择全量基因库的概率为[g/100],其中g表示种群的代数。突变基因的概率分布由基因MIC分数的分布渐变为均匀分布。
2.4 移除重复个体
上述流程会产生大量重复的个体,虽然个体的重复有一定的概率意义,即优势个体的重复多,保留下来的概率大,但其极大影响了种群的丰富度,还会导致算法过早收敛陷入局部最优解当中。
图4是使用种群个体去重操作前的结果图,左图表示的是基因重复数排名前10的基因占到种群基因总数的比例, 在一定程度上反映了种群中基因的集中程度,右图是每代种群中基因的种类数,反映了种群中基因的丰富程度,由上图我们可以发现,基因的丰富度减小的极快而且非常集中,基因的丰富度小,算法的随机性差,很容易陷入局部最优解而过早收敛。
图5是采用个体去重的结果图。可以发现由于在每一代个体都进行了去重,重复基因数量排名前10的基因,基因总数最多只占到基因总数的60%左右,基因的种类也维持在100个左右,由于基因的丰富度增大了,算法的随机性便增强了,有效地避免了算法收敛到局部最优解。
3 实验结果
3.1 实验数据集
实验使用了5种不同的基因芯片公开数据集来验证本算法的有效性,数据集包含了二分类数据集和多分类数据集,其中二分类数据集包括colon,carcinoma,leukemia数据集,其中colon和carcinama包含两个类别,一个是患病类,一个是正常类,leukemia则包含两个患病类,他们分别是急性淋巴细胞白血病(ALL)和急性髓性细胞白血病(AML)。多类别数据集有SRBCT和MLL,其中SRBCT数据集包含4种癌症类别,他们分别是尤文氏肉瘤(EWS),非霍奇金淋巴瘤(NHL),神经母细胞瘤(NB)和横纹肌肉瘤(RMS),MLL数据集包含3种类别,分别是急性淋巴细胞白血病(ALL),急性髓性细胞白血病(AML)和混合血统白血病(MLL)。
3.2 实验结果分析
在各数据集上独立重复进行20次,与其他算法相对比,结果见表1。
4 结论
本文从选择算子,变异算子和种群个体冗余入手,改进了遗传算法挑选特征基因和变异的方法。通过实验得出,仅改变选择和变异算子,算法准确率有所提升,考虑种群去重和选择算子之间的相互作用时,准确率相对差一些,这是后续的研究中需要解决的一个问题。
参考文献:
[1] 王翔,胡學钢.高维小样本分类问题中特征选择研究综述[J]. 计算机应用,2017(9):2433-2438.
[2] 刘金勇.基因表达谱数据特征选择与提取方法研究[D].杭州:中国计量学院,2014.
[3] 翟云,杨炳儒,曲武. 不平衡类数据挖掘研究综述[J]. 计算机科学, 2010, 37(10):27-32.
[4] 于化龙, 高尚, 赵靖, 等. 基于过采样技术和随机森林的不平衡微阵列数据分类方法研究[J]. 计算机科学, 2012(5):196-200.
[5] 陈岩, 来海锋, 王清, 等. 基于filter-wrapper的两步特征变量提取方法[J]. 机电工程, 2010(4): 67-71.
[6] Li S , Wu X , Hu X . Gene Selection Using Genetic Algorithm and Support Vectors Machines[J]. Soft Computing, 2008, 12(7):693-698.
[7] Motieghader H, Najafi A, Sadeghi B, et al. A Hybrid Gene Selection Algorithm for Microarray Cancer Classification Using Genetic Algorithm and Learning Automata[J]. Informatics in Medicine Unlocked, 2017: 246-254.
[8] 胡局新, 张功杰. 基于K折交叉验证的选择性集成分类算法[J]. 科技通报, 2013(12):123-125.
【通联编辑:唐一东】
