高效逆流色谱法制备苏木中的氧化巴西木素
2020-11-02和文倩范青飞黄凤梅胡华斌宋启示
和文倩, 范青飞, 周 兰, 黄凤梅, 蒋 仙, 纳 智, 胡华斌*, 宋启示*
(1. 中国科学院西双版纳热带植物园, 热带植物资源可持续利用重点实验室, 云南 昆明 650223; 2. 中国科学院大学, 北京 100049; 3. 普洱学院, 云南 普洱 665000)
氧化巴西木素是苏木主要成分之一。它是由巴西苏木素在日光照射下和空气中的氧气发生氧化反应生成的[1]。现代药理研究显示,氧化巴西木素具有抗肿瘤[2,3]、抗炎[4]、抗菌[5]、抗氧化[6]和调节免疫[7]等多种药理活性。此外,氧化巴西木素作为苏木天然色素成分之一,被广泛应用于食品、日化、皮革及织物染色等行业[8],在病理实验中还被用作细胞组织切片的染色剂[9]。氧化巴西木素应用前景相当广阔,加大力度开发其制备工艺,将会对现代医疗及染色工业的发展起着极大帮助。
目前,主要采用传统柱色谱方法对氧化巴西木素进行分离纯化,氧化巴西木素主要集中在乙酸乙酯部位,借助大孔树脂色谱、硅胶柱色谱、葡聚糖凝胶色谱、高效液相色谱对苏木的乙酸乙酯部位进行分离纯化[10-12]。本研究组在应用传统柱色谱方法从苏木中分离巴西苏木素及氧化巴西木素时,发现色谱柱中的固体填充材料不仅被染成红色,而且红色难以被洗脱,从而造成色谱柱材料的污染以及样品成分的损失。高效逆流色谱(HPCCC)是一种新型的液液分配萃取技术,无须固体支撑材料,可避免样品被吸附或损失。高效逆流色谱分离速度快,工作效率高,能够克服传统分离方法中存在的缺点,非常适合用于分离具有特殊性质的天然产物活性成分[13-16]。
本研究利用HPCCC对苏木中的活性成分进行分离纯化。通过优化溶剂系统对氧化巴西木素进行快速分离纯化,同时获取一微量成分,并对其进行纯度分析和结构鉴定。该方法不仅快速,简便,还可避免苏木中的活性成分氧化巴西木素对色谱柱中的固体填充材料染色、难以洗脱等问题。
1 实验部分
1.1 仪器、试剂和材料
分析兼半制备型高效逆流色谱仪(Spectrum HPCCC,英国Dynamic Extractions公司),分析高效液相色谱仪(e2695,美国Waters公司),恒流输液泵、检测器(德国Knauer公司),循环水冷却器(SH150-1000,美国Lab Tech公司),电子天平(中国上海梅特勒-托利多仪器有限公司),超声清洗器(中国上海Sapeen科学仪器有限公司),核磁共振仪(Bruker AM-400/500 MHz,瑞士Bruker公司)。
HPCCC溶剂系统所用试剂氯仿、甲醇、石油醚、乙酸乙酯为工业重蒸,水是实验室自制纯净水,HPCCC分析用色谱甲醇(德国Merck公司),二甲基亚砜-d6(DMSO-d6, 中国百灵威科技公司,包含0.03% (v/v)四甲基硅烷(TMS))。
实验植物材料苏木由云南西双版纳州傣医院提供,经鉴定为苏木心材。
1.2 实验方法
1.2.1样品前处理
将苏木干燥样品(10 kg)粉碎,然后按照料液比1∶5 (kg/L)加入50 L 95%乙醇热回流提取3次,合并提取液,减压浓缩并旋干,得到乙醇提取物600 g。按照料液比1∶2加蒸馏水溶解,依次用石油醚、乙酸乙酯、正丁醇溶剂对其进行多次萃取,得石油醚萃取物4.84 g、乙酸乙酯萃取物494.2 g、正丁醇萃取物48.6 g,称取乙酸乙酯萃取物500 mg进行逆流分离制备。
1.2.2HPCCC分离过程
以氯仿-甲醇-水(4∶3∶2, v/v/v)为溶剂体系,充分混合后,静置分层,上相溶剂为固定相,下相溶剂为流动相。以流速15 mL/min将固定相泵满整个螺旋管柱(300 mL),启动主机,调整主机转速为1 600 r/min,在15 min左右达到基本稳定,此时以流速10 mL/min泵入流动相。待流动相从主机出口流出时,注入已过滤好的样品,并用自动馏分收集器收集样品。主机温度控制在25 ℃,检测波长设置为285 nm,分析扫描时间设置为120 min。
1.2.3HPLC分析条件
将苏木乙酸乙酯提取物和通过高效逆流分离出的组分进行HPLC的纯度分析,样品用甲醇溶解后用过滤头装入2 mL带盖液相试剂瓶中。采用分析型Symmety®C_18色谱柱(250 mm×4.6 mm, 5 μm,美国Waters公司),梯度梯度洗脱模式,二极管阵列检测器(PDA)全波长检测,以甲醇(A)和水(B)作为流动相。流动相洗脱程序为0~30 min, 5%A~95%A; 30~35 min, 95%A; 35~40 min, 95%A~5%A。流速0.8 mL/min,波长285 nm。
2 结果与讨论
2.1 待测样品分析
苏木的活性部位主要集中在乙酸乙酯部分[17,18]。根据以往的经验,需对苏木乙酸乙酯萃取物进行HPLC分析,根据其保留时间来推算极性大小。苏木乙酸乙酯萃取物HPLC分析结果见图1,其大部分化合物出峰时间较早(12~20 min)。根据实验设计,将图1中一个峰设定为目标峰并标记,通过洗脱时间可以推断该目标峰的化合物在乙酸乙酯部位极性相对较大。
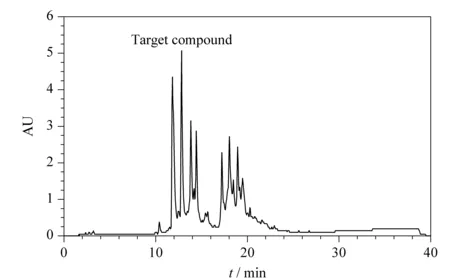
图 1 苏木乙酸乙酯萃取物的HPLC色谱图Fig. 1 HPLC chromatogram of the ethyl acetate extract of Caesalpinia sappan
2.2 溶剂体系的筛选
在逆流色谱中,溶剂系统的选择至关重要。很多研究者成功应用逆流色谱技术对不同类型化合物实现了分离[19,20]。大部分研究者在进行溶剂系统筛选时会考察目标化合物的分配系数,接着研究所用逆流色谱的参数,如转速、固定相保留率等对分离效果的影响[21-23]。本文在无须计算目标化合物的分配系数的前提下,结合本研究组的工作经验及化合物特点,利用基于薄层色谱的常用溶剂体系估算法(TLC-GUESS法)和摇瓶法(Shake-Flash法)结合高效逆流色谱分析模式进行溶剂体系的快速筛选。具体的筛选过程如下:
TLC-GUESS方法由Friesen教授和Pauli教授建立,成功应用于逆流色谱溶剂系统的选择[24]。首先选择常规溶剂系统即石油醚-乙酸乙酯-甲醇-水系统,通过TLC-GUESS法分析该溶剂系统的可行性,调整溶剂比例后,选定适合的溶剂比例,并通过分析逆流色谱进行验证。结合课题组之前的工作[25],我们通过TLC-GUESS方法选择溶剂比例为石油醚-乙酸乙酯-甲醇-水(3∶7∶3∶7, v/v/v/v),并应用逆流色谱分析模式对该溶剂体系进行验证,从图2a可以看出,在该溶剂条件下,大部分化合物保留时间较短,峰出现重叠现象,不能实现化合物分离。

图 2 两种溶剂体系分离苏木乙酸乙酯萃取物的HPCCC色谱图Fig. 2 High performance countercurrent chromatography (HPCCC) chromatograms of the ethyl acetate extract of Caesalpinia sappan with two solvent systems a. petroleum ether-ethyl acetate-methanol-water (3∶7∶3∶7, v/v/v/v); b. chloroform-methanol-water (4∶3∶2, v/v/v).
氧化巴西木素为黄酮类化合物,查阅相关资料[26],尝试使用氯仿-甲醇-水溶剂体系。准备常用的氯仿-甲醇-水(4∶3∶2, v/v/v)溶剂体系,将样品溶解于等体积的上下相溶剂中,剧烈摇晃5 min后静置,待上下相分层后可观察样品上下相的颜色深浅,判断上下相分布是否均匀,进而判断该溶剂系统是否适合做备选溶剂系统(Shake-Flash法)。经实验发现,样品上下相颜色深浅一致、分布均匀,即可判断氯仿-甲醇-水(4∶3∶2, v/v/v)溶剂体系适合做备选溶剂系统,应用逆流色谱分析模式对该溶剂系统进行验证(见图2b)。
在溶剂系统中,目标物的分配系数(K)一般在0.5~2.0之间,如果大于2.0或小于0.5,目标化合物可能滞后或提前被洗脱。在TLC-GUESS方法实际操作中,如果一个样品中的化合物既有分布在0.5~2.0之间的,也有分布在该区间两侧的,则认为该溶剂系统是比较合适的。通过比较分析逆流色谱图2a和图2b,发现图2b中的化合物出峰较均匀,适合化合物分离,最终,氯仿-甲醇-水(4∶3∶2)溶剂系统被选做较佳溶剂体系。
2.3 HPCCC的制备
本实验选择氯仿-甲醇-水(4∶3∶2, v/v/v)作为HPCCC分离苏木乙酸乙酯提取物的溶剂系统。样品上样量为500 mg,应用反相洗脱模式对样品进行分离(见图3)。苏木中含有化合物种类较多,各个峰之间分离度较小,运用自动收集器对样品进行收集。通过TLC点板分析,发现馏分ccc-32和ccc-39仅有一个主点。故采用1.2.3节的HPLC法对HPCCC收集到的ccc-32和ccc-39进行纯度分析并进行鉴定(见图4)。根据保留时间,峰面积归一化法推断馏分ccc-39为化合物1(15.2 mg,纯度为95.6%), ccc-32为化合物2(5.3 mg,纯度为89.0%)。组分ccc-32与图1中的苏木乙酸乙酯萃取物没有较明显重合的峰,推断组分化合物2为苏木乙酸乙酯萃取物中一微量成分。可见逆流色谱技术可以应用于微量成分的富集和分析中。
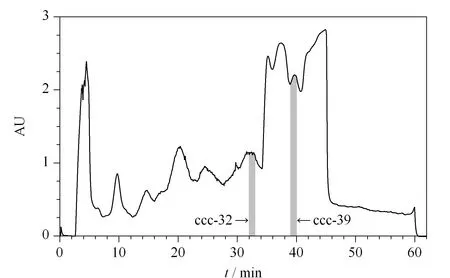
图 3 苏木乙酸乙酯萃取物的高效逆流色谱图Fig. 3 HPCCC chromatogram of the ethyl acetate extract of Caesalpinia sappan

图 4 HPCCC分离得到的ccc-39和ccc-32的HPLC色谱图Fig. 4 HPLC chromatograms of ccc-39 and ccc-32 from the ethyl acetate extract of Caesalpinia sappan by HPCCC
2.4 化合物的结构鉴定
将分离的化合物进行1H-NMR和13C-NMR分析,对其进行结构鉴定。具体数据如下:
化合物1:分子式C16H12O5,红色无定型粉末;1H-NMR (500 MHz, DMSO-d6)δ: 7.79 (1H, d,J=8.9 Hz, H-1), 7.10 (1H, s, H-11), 6.56 (1H, dd,J=8.9, 2.4 Hz, H-2), 6.35 (1H, d,J=2.4 Hz, H-4), 6.31 (1H, s, H-8), 4.44 (1H, d,J=11.6 Hz, H-6), 4.00 (1H, d,J=11.8 Hz, H-6), 2.83 (1H, d,J=18.4Hz, H-7), 2.81 (1H, d,J=18.4 Hz, H-7′);13C-NMR (125 MHz, DMSO-d6)δ: 179.4 (C-9), 162.3 (C-3), 159.0 (C-7a), 157.7 (C-4a), 152.4 (C-10), 151.6 (C-12), 130.6 (C-1), 126.1 (C-11a), 117.5 (C-8), 110.8 (C-1a, 2), 104.1 (C-11), 102.9 (C-4), 74.2 (C-6a), 72.9 (C-6), 39.7 (C-7)。以上数据与文献[18]基本一致,故鉴定为氧化巴西木素,化学结构如图5所示。

图 5 化合物1和2的结构式Fig. 5 Chemical structures of compounds 1 and 2
化合物2:分子式C32H32O12,1H-NMR (800 MHz, DMSO-d6)δ: 6.91 (2H, d,J=8.0 Hz, H-12), 6.91 (2H, d,J=8.0 Hz, H-12′), 6.68 (1H, s, H-6), 6.62 (1H, s, H-3), 6.59 (1H, s, H-6′), 6.54(1H, s, H-3′), 6.47 (1H, dd,J=8.8 Hz, H-11′& 11), 6.46 (1H, d,J=2.2 Hz, H-9′), 6.45(1H, dd,J=2.2 Hz, H-9), 4.25 (1H, d,J=12 Hz, H-15′), 4.13(1H, d,J=12 Hz, H-15), 3.95 (1H, d,J=12 Hz, H-15), 3.93 (3H, m, H-15′, 16, 16′), 3.75 (1H, d,J=12 Hz, H-16′), 3.73 (1H, d,J=12 Hz, H-16), 4.25 (1H, d,J=12.0 Hz, H-16′), 3.74(1H, d,J=12.0 Hz, H-16), 2.68 (2H, s, H-13′), 2.37 (1H, d,J=13.6 Hz, H-13), 2.34(1H, d,J=13.6 Hz, H-13);13C-NMR (200 MHz, DMSO-d6)δ: C 159.2 (C-8), 157.8 (C-8′), 157.7 (C-10′), 157.6 (C-10), 143.6 (C-4, 4′), 143.6 (C-5), 143.6 (C-5′), 131.9 (C-12), 131.0 (C-12′), 131.0 (C-1), 129.5 (C-1′), 126.8 (C-2′), 126.3 (C-2), 123.8 (C-7′), 121.9 (C-7), 119.4 (C-3), 118.4 (C-3′), 116.6 (C-6′), 116.1(C-6), 111.0 (C-11′), 110.1 (C-11), 107.9 (C-9′), 106.9 (C-9), 76.3 (C-15′), 74.7 (C-15), 71.4 (C-14′), 71.2 (C-14), 67.2 (C-16), 64.3 (C-16′), 42.0 (C-13′), 38.9 (C-13)。以上数据与参考文献[27]基本一致,故鉴定为caesappanin C,化学结构见图5。
3 结论
氧化巴西木素不仅是一药用化学成分,还是天然的染色剂,具有染色功能,用传统的柱色谱法分离,会将色谱柱中的固体填充材料染成粉红色且难以洗脱,造成对色谱柱材料的污染以及氧化巴西木素的损失。本文应用HPCCC对苏木中的活性成分氧化巴西木素进行了快速分离制备。该方法简便可行,不仅可以解决氧化巴西木素的染色污染的问题,而且还可以为类似染色类成分分离制备提供技术指导。
