MicroRNA sequences modulating inflammation and lipid accumulation in macrophage “foam” cells:Implications for atherosclerosis
2020-10-31RichardJamesLightbodyJaniceMarieWalshTaylorYvonneDempsieAnnetteGraham
Richard James Lightbody, Janice Marie Walsh Taylor, Yvonne Dempsie, Annette Graham
Abstract
Key words: Coronary heart disease;Atherosclerosis;Macrophage “foam” cell;Cholesterol;Inflammation;MicroRNA
INTRODUCTION
The purpose of this review is to identify and contextualise the emerging roles of micro-RNA (miRNA) sequences involved in epigenetic regulation of cholesterol deposition within macrophage “foam” cells, a rapidly developing area of key interest to researchers and clinicians developing new therapeutic strategies to combat coronary heart disease (CHD).CHD, a major cause of global morbidity and mortality, is principally caused by atherosclerosis, a complex, progressive chronic inflammatory disease.Genetic factors contribute to atherosclerosis, in combination with environmental, metabolic and behavioural triggers including elevated serum lipid levels, diabetes, obesity, hypertension and smoking[1].Atherosclerotic lesions originate at non-random locations of the vasculature[2,3], where alterations in haemodynamic blood flow, such as decreased shear stress and turbulent flow, are sensed by endothelial cells, disrupting homeostatic cellular organisation, increasing permeability of the arteries and enabling the accumulation of circulating cholesterol-rich lowdensity lipoprotein (LDL) in the intima[2-4].Local inflammation in endothelial cells is mediated by activation of the pro-inflammatory transcription factor, nuclear factor-κB(NF-κB), in part due to shear stress-mediated inhibition of the anti-inflammatory transcription factor Kruppel-like factor 2[5,6].This leads to increased expression of adhesion molecules, E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1, and of pro-inflammatory cytokines and chemoattractants[6].Further, endothelial expression of lipoxygenase enzymes and production of reactive oxygen species (ROS) oxidatively modify proteoglycan-bound LDL (oxLDL)[7].This amplifies the local inflammatory response mediated by receptors such as lectin-like oxidised LDL receptor 1 (LOX-1) and toll-like receptor 4 (TLR4)present on endothelial and smooth muscle cells[8-10].
Endothelial expression of adhesion molecules, and chemokine-chemokine receptor interactions, recruit circulating monocytes to the intima, where they differentiate into macrophages in response to macrophage colony-stimulating factor[11].Intimal macrophages recognise modified components of oxLDL and internalise oxLDL and LDL, becoming “foam” cells due to accumulation of droplets of cholesteryl ester.This occursviainteraction with scavenger receptors (SRs), such as SR-A1, SR-B1, cluster of differentiation 36 (CD36, SR-B2), CD68 (SR-D1), LOX-1 (SR-E1), TLR4, and the LDL receptor (LDLR)[7,12-14].Lipoprotein lipase (LPL) is also implicated in foam cell formation in distinct ways:Inhibition of LPL activity by angiopoietin-like protein 4 decreases lipid uptake in macrophages, whereas genetic deletion of this protein increases lipid uptake, expression of lipid-induced genes and respiration[15].Additional receptor independent mechanisms such as macro- and micropinocytosis can also lead to the uptake of these lipoproteins[16,17].The influx of cholesterol-rich lipoproteins through these various mechanisms, as well as the rate limited process of cholesterol efflux (below), leads to the generation of lipid-laden “foam” cells with reduced capacity to migrate from the intima[18-20].
Accumulation of macrophages and lipid-laden foam cells is accompanied by plaque enrichment with additional immune cells[21].T-helper cells, activated by oxLDLinduced maturation of dendritic cells (DCs), recognise epitopes on apolipoprotein B100 (ApoB100) in native LDL and oxLDL[22,23].Phenotypically, these THcells are primarily of the TH1 subset, producing pro-inflammatory cytokines such as interferon gamma (IFN-γ) and tumour necrosis factor alpha (TNF-α), but atheroprotective antiinflammatory regulatory T cells (Tregs) are also present[24,25].The combination of hyperlipidaemia, endothelial expression of adhesion molecules and chemokines, and deposition of chemokines on endothelial cells by activated platelets, recruits and activates additional immune subsets, including neutrophils[26-30].Neutrophils express myeloperoxidase that produces hypochlorous acid, that promotes LDL oxidation and foam cell formation and increases retention of LDL in the intimaviabinding to LPL[7,26-31].
As the complexity of the arterial microenvironment increases, atherosclerotic plaques develop a number of key features.Responding to signals such as growth factors, cytokines and oxidised phospholipids (oxPL), vascular smooth muscle cells(VSMCs) undergo phenotypic switching from contractile quiescent VSMCs to synthetic, migratory and proliferative VSMC[32,33].This leads to dramatic vascular remodelling and arterial thickeningviaproduction of matrix degrading metalloproteinases and a shift in production from type I and III collagen to type VIII collagen[34-36].Further, intimal VSMCs accumulate lipids and can take on a foam cell phenotype which, under endoplasmic reticulum (ER) stress or in the presence of increased intracellular free cholesterol, leads to apoptosis and necrosis of both macrophage-derived and VSMC-derived foam cells, forming a hypoxic, necrotic core and extracellular lipid pools[37-41].Hypoxia inducible factor 1 enhances neovascularisationviainduction of vascular endothelial growth factor A expression in macrophages and VSMC[42,43], and by increased expression of macrophage SRs and proinflammatory mediators and decreased expression of ATP binding cassette (ABC)transporters responsible for cellular cholesterol efflux[44-46].
Macrophage sub-populations within atherosclerotic lesions
Within the arterial intima, macrophages exhibit notable phenotypic plasticity in response to multiple signals from this complex microenvironment and can exhibit proor anti-atherosclerotic responses (Figure 1).Pro-inflammatory (M1) macrophages can be generatedin vitroin response to a variety of stimuli associated with a TH1 response,such as lipopolysaccharide (LPS) and IFN-γ[47,48], resulting in increased expression of pro-inflammatory mediators such as interleukin (IL)-1β, IL-6, TNF-α, IL-12 and IL-23,and ROS[47,49,50].Oxidized LDL induces activation of the NF-κB pathway which enhances the pro-inflammatory response in M1-like macrophages and expression of pro-inflammatory mediators in macrophages polarised to the anti-inflammatory (M2)phenotype[51-53].Further, oxLDL and individual components derived from oxLDL, such as free cholesterol and cholesterol crystals, cholesteryl ester hydroperoxides, and 7-ketocholesteryl-9-carboxynonanoate (Figure 1), have been shown to activate proinflammatory pathways including the nod-like receptor protein 3 (NLRP3)inflammasome[54], mitogen activated protein kinase pathway[55,56]and NF-κB signalling[57,58].
Macrophages, however, can adopt a variety of additional immunoregulatory subtypes within the phenotypic spectrum.A subset of anti-inflammatory macrophages(M2a), generated in response to cytokines such as IL-4 and IL-13, are produced as part of the TH2 response[59,60].These cells are associated with wound healingviaproduction of factors such as fibronectin and transforming growth factor β (TGF-β)[59,60].Exposure to immune complexes, and to TLR ligands, generates another subset, termed M2b,which have both protective and detrimental roles and express high levels of the antiinflammatory cytokine IL-10, low levels of pro-inflammatory IL-12, but also other proinflammatory mediators including chemoattractant C-C motif chemokine ligand 1(CCL1)[61-63].Stimulation of macrophages with glucocorticoids and IL-10 induce macrophage phenotype (M2c), which play a predominant role in clearance of apoptotic cells[64,65].Finally, a pro-angiogenic population, termed M2d, is induced in murine macrophages in response to adenosine agonists in conjunction with TLRsignalling[66,67].
Interestingly, while some components of oxLDL activate inflammatory pathways,oxysterols and the product of cholesteryl ester oxidation, 9-oxonanoyl cholesterol,induce expression of the anti-inflammatory and pro-fibrotic cytokine TGF-β[68,69].Consequently, treatment of macrophages with oxLDL can also induce an antiinflammatory phenotype[70].This apparent discrepancy between inflammatory versus anti-inflammatory signalling may be due to the degree of oxidation of the LDL particle[71], or the extent of lipid accumulation within cells[72].Oxidized phospholipids(oxPL) (Figure 1) also induce a distinct macrophage phenotype (Mox) in murine models[73-75];Mox macrophages exhibit reduced expression of M1, as well as M2-like,macrophage markers and enhanced expression of nuclear factor erythroid 2-related factor 2 (Nrf2) dependent anti-oxidant genes[73].Further, macrophage subsets found within the plaque microenvironment include M4 macrophages, which are induced by chemokine (C-X-C) motif ligand 4 (CXCL4)[76], MHb macrophages, induced by hemoglobin:Haptoglobin complexes[77], and Mhem macrophages, induced by heme[78,79];both MHb and Mhem macrophages exhibit resistance to cholesterol loading[77-79].

Figure 1 Macrophage phenotype in response to lipids and other factors within atherosclerotic lesions.
Cholesterol accumulation in macrophage “foam” cells:Responses to excess (oxy)sterol
Macrophages play a vital role in handling excess LDL-cholesterol and/or toxic oxLDL metabolites,viaesterification to cytosolic lipid droplets by Acyl CoA:Cholesterol Acyltransferase (ACAT-1-/2) or lysosomal sequestration[80].Autophagy also contributes to lipid droplet formation, with Beclin-1 inhibiting the formation of droplets in response to modified LDL in naïve cells but not in inflammatory activated macrophages[81].Accumulation of sterol is central to activation of nuclear liver X receptors (LXRs), which lie under the control of oxysterol metabolites of cholesterol;the extent of activation of these transcription factors may also contribute to the heterogeneity of macrophage responses to sterol accumulation[72,82].LXRs control the expression of ABC transporters (ABCA1, ABCG1/G4) that actively efflux cholesterol from cells to acceptors, such as apolipoprotein (apo) A-I, apoE and high-density lipoprotein (HDL).Binding of apoA-I to ABCA1 triggers an array of cell signalling pathways[83], and mobilisation of stored cholesteryl estersviacholesteryl ester hydrolases, releases cholesterol which trafficks to the plasma membrane for efflux as nascent HDL[80,84].In addition, ABCG1 and ABCG4 aid the formation of more mature forms of HDL, so they work in concert with ABCA1 to initiate the process of reverse cholesterol transport which can return cholesterol to the liver for excretionviathe classical and alternative bile acid pathways[80,84].
Liver X nuclear receptors form obligate heterodimers with retinoid X receptors which bind directly to the LXR response element, a direct repeat 4 (DR4) motif of the six base pair sequence AGTTCA separated by four base pairs[85-88].Ligand binding triggers a conformational change in the heterodimer, dissociating nuclear receptor corepressors [NCOR1/NCOR2 (SMRT)] proteins which undergo ubiquitination and proteasomal degradation, and engaging coactivator proteins (steroid receptor coactivators, PPAR-γ coactivator 1 and nuclear receptor coactivator 6)[87].Genetic deletion of LXRs in bone marrow derived macrophages, however, reveal a more complex picture:Depending on the target gene, LXR deletion can up- or downregulate, or effect no change in, gene expression[87].
LXRs contribute to the inactivation of the counter-regulatory system operated by sterol regulatory binding proteins (SREBPs) which belong to the basic helix-loop-helix leucine zipper (bHLH-Zip) family of transcription factors[89].Three SREBP isoforms exist, encoded by two genes:SREBF1(SREBP-1a and SREBP-1c) andSREBF2(SREBP-2) which target sterol response elements (SRE).Unlike SREBP-1a which is constitutively expressed and targets all SRE with low specificity, SREBP-1c and SREBP-2 are inducible and regulate the expression of genes encoding proteins involved in fatty acid and cholesterol metabolism, respectively[89,90];SREBP-2 also transcriptionally regulates the LDL receptor which mediates endocytosis of LDL from the circulation.In sterol-replete cells, SREBP transcription factors remain inactive,sequestered at the ER by binding to a chaperone, SREBP cleavage activating protein(SCAP), which contains a five transmembrane sterol sensing domain and interacts with the ER anchor, insulin-induced gene (INSIG-1/-2).As cholesterol levels fall, the interaction of SCAP with INSIG is lost, allowing SCAP-SREBP to traffick to the Golgi apparatusviainclusion in COPII-vesicles[89,90].Golgi site-1 and site-2 proteases (SP-1,SP-2) cleave the amino terminal of SREBP-2, releasing a transcriptionally active fragment which is imported into the nucleus to target sterol-responsive genes,includingSREBF2itself;rapid degradation of nuclear (nSREBP) serves to terminate this signalling pathway[89,90].
LXRs operate in functional opposition to SREBP-2, repressing cholesterol biosynthesisvianovel negative LXR DNA-response elements in the promoter region of genes encoding squalene synthase and lanosterol 14-demethylase[91-93], and promoting the degradation of LDL receptors by increasing the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9)[94,95].The E3 ubiquitin ligase, inducible degrader of the LDL receptor (IDOL) is an LXR target gene:IDOL dimers interact with members of the ubiquitin-conjugating enzyme (UBE) 2D family of E2 ubiquitin ligases to transfer ubiquitin to the cytoplasmic tail of members of the LDL receptor family,promoting receptor degradation[94,95].Oxysterols also bind to Insig-1/2, sequestering SREBPs at the ER, further ensuring repression of cholesterol biosynthesis and uptake[88].By contrast, LXR agonists induce gene expression of SREBP-1c;fatty acid synthase, and a number of desaturase and elongase enzymes in the fatty acid biosynthetic pathway, are also directly regulated by LXR[87].Thus, efficient delivery of cholesterol to the ER is needed to inhibit proteolytic processing of SREBP-1c to an active transcription factor[89,90], and to limit increased biosynthesis of fatty acids[87].
LXRs also exert anti-inflammatory effects, some of which are indirect and due to the increased expression of ABCA1[96], and production of anti-inflammatory HDL[97,98].HDL inhibit TLR signalling in macrophages and cytokine signalling in bone marrow progenitors by removal of cholesterol from lipid rafts[99,100]and induce activating transcription factor 3, suppressing the expression of pro-inflammatory genes[97,98].Multiple mechanisms exist by which LXRs modulate inflammatory responses, some of which involve transactivation and others transrepression[86-88].Pathway-specific responses occur:LXR activation inhibits NF-κB dependent induction of proinflammatory genes in response to LPS and responses triggered by TLR4 and TNF-α but exerts minimal impact on the pathway mediated by TLR3[86-88].LXRs also regulate apoptosis and enhance survival of macrophages within lesions, while IFN-γ promotes neointimal hyperplasia and macrophage apoptosis by promoting ubiquitin-dependent LXR degradation[101,102].
However, it should be recognised that the pharmacology of oxysterols is highly complex:A large number of nuclear, and G-protein coupled (GPR), receptors bind these bioactive lipids [e.g., retinoid-related orphan receptors, ER, Epstein-Barr virus induced GPR (EBI2/GPR183) and IL-8 receptor (CXCR2)][93].This, combined with the complexity of oxysterol metabolism, enzymatic conversion to other species such as esters, bile acids and 3-sulphate derivatives, and tissue- and species-specific effects,makes deciphering the (patho)physiological impact of these molecules particularly challenging[93].
EPIGENETIC MECHANISMS CONTRIBUTING TO “foam cell” FORMATION:THE EMERGING ROLE OF MICRORNA
It is increasingly clear that epigenetic mechanisms such as DNA methylation, histone post-translational modification and changes in expression of non-coding RNA, such as long non-coding RNA (lncRNA) and miRNA, are important contributors to macrophage phenotype and the pathogenesis of “foam cell” formation.Alterations in chromatin structure and gene expression exert both acute and chronic effects on a wide array of biological processes which influence macrophage lipid accumulation and inflammatory responses.For example, DNA methyltransferases catalyse methylation of the 5’-position of cytosine residues, using S-adenosyl-methionine as the methyl donor, resulting in hypermethylation of CpG islands and stable repression of transcription[103,104].Chromatin histone post-translational modifiers, such as histone acetyltransferases and deacetylases and histone methyltransferases, target lysine and/or arginine residues to induce or repress gene expression, dynamically finetuning gene expression by controlling the access of transcription factors to promoter and enhancer regions[105,106].
An additional layer of epigenetic regulation is provided by non-coding RNA sequences, including lncRNA sequences, longer than 200 nucleotides, and miRNA sequences (20-25 nucleotides in size), the focus of this review article, which fine-tune expression of multiple (networks of) genes in response to environmental factors,including oxLDL[107-109].Sequences encoding miRNA can be found singly or in clusters throughout the genome, located in intron-exon portions of protein-encoding genes or intergenic regions[110,111].Transcription is dependent on the activity of RNA polymerase II/III and expression, in relation to intergenic miRNA, can be dependent or independent of host gene expression[111-114].MicroRNA are frequently found in clusters and can be co-transcribed and separated by splicing, or expressed independently[115].Transcription and generation of miRNA occurs through both canonical and noncanonical pathways, with less information available on the latter[116].The canonical pathway involves generation of a hairpin-containing primary miRNA (pri-miRNA)transcript containing a 5’ methylated cap and a 3’ polyadenylated tail required for primiRNA processing and transport[112,117].Processing occursviaa microprocessor complex consisting of the double-stranded RNA-binding protein DiGeorge syndrome critical region gene 8 which recognises methyl motifs present in the pri-miRNA[118,119].This interaction serves as an anchor for a ribonuclease II (RNase III), known as Drosha,which cleaves the hairpin structure from the pri-miRNA transcript generating precursor miRNA (pre-miRNA)[120-122].
Export of pre-miRNA (around 70 nucleotides in length) from the nucleus involves the nucleocytoplasmic transporter factor exportin-5 and Ras-related nuclear protein(Ran)GTP[123].Recognition and binding to exportin-5 occurs primarily through interaction with the 3’ overhanging sequence of pre-miRNA.Blunt ended pre-miRNA remain capable of interaction while RanGTP is bound to the hairpin structure,following release into the cytoplasm[124,125];hydrolysis of GTP to GDP results in release of the pre-miRNA[118].Once localised in the cytoplasm, pre-miRNA is processed by a second RNase III enzyme, Dicer, to a mature miRNA duplex (19-25 nucleotides)through removal of the stem-loop structure[126,127].The guide strand, which has lower base pairing stability, is loaded onto the RNA-induced silencing complex (RISC)composed of Dicer, transactivation response (TAR) RNA binding protein (TRBP) and Argonaute proteins (1 to 4).After integration into the active RISC complex, miRNAs base pair with their complementary mRNA molecules, guided by their miRNA recognising element[128,129].
Degradation of target mRNA occurs only when the miRNA and the target mRNA match exactly (perfect match) or are nearly exactly complementary to each other;this process is the same as the RNA interference induced by artificial small interfering RNA (siRNA)[130,131].By contrast, if the complementarity between miRNA and target mRNA is only partial (imperfect match), then more moderate reductions in mRNA levels accompanied by translational repression will occur.Targeting occurs through binding of the seed sequence of RISC-incorporated miRNA to conserved complementary regions found in the 3’UTR of target mRNA[130,131].Factors such as AUrich regions near seed region binding sites, and auxiliary binding of the miRNA to transcript can also play a role in determining target specificity, reducing translational efficiency or inducing mRNA destabilisationviadeadenylation[132-134].miRNA also exert regulatory functions on gene expression in the nucleus, paradoxically promoting gene expression in certain conditions[135,136].In eukaryotic cells, miRNA molecules can bind several target sequences, mainly within the 3’-UTR of mRNA with varying degrees of complementarity, so that each single miRNA is able to interact with and regulate a large number of genes.Computational prediction suggests that more than 60% of all mammalian protein-coding genes are conserved targets of miRNA, while each miRNA has target sites in hundreds of different genes[137,138];miRNAs also display tissuespecific expression[139]and concentration-dependent effects in pathologically affected organs and tissues[140-142].
miRNA not only regulate the transcriptional landscape of the cell, but some sequences exist in the extracellular environment in a variety of different forms;degradation of miRNA is avoided through association with Argonaute RISC catalytic component 2 (Ago2), and to a lesser extent nucleophosmin 1 (NMP1)[143-145].miRNA are found enriched in extracellular vesicles such as exosomes, microvesicles, and lipoproteins such as HDL[146-148], and represent novel biomarkers of atherosclerosis[149].Secreted miRNA may elicit pro- and anti-atherosclerotic functions:EC when placed under conditions of atherogenic shear stress release Ago2-bound miR-126-3p which in turn downregulate contractile VSMC markers[150], while delivery of miR-223 by HDL to EC leads to downregulation of ICAM-1[151,152].
miRNA sequences implicated in macrophage “foam” cell formation
Over the last five years, there has been an explosion of interest in the role of miRNA involved in macrophage biology, and in “foam” cell formation in particular.Table 1[153,155-204]indicates some of the miRNA sequences identified by interrogation of the NCBI PubMed database, as either altered by uptake of modified LDL by macrophages,or implicated in the pathogenesis of foam cell formation.Many of the genes targeted by these sequences play established roles in either lipid metabolism or inflammation,but a significant number have no prior links to either process, highlighting the importance of miRNA research in driving the discovery of novel cellular processes contributing to disease.

Table 1 MicroRNA sequences associated with macrophage ”foam cell” formation and atheroma

Lv et al[163], 2015 Liang et al[164], 2017 Feng et al[165], 2014 Canfrán-Duque et al[166], 2017 Yang et al[167], 2018 Di Gregoli et al[168],2014 Zhang et al[169], 2014 Li et al[170], 2018 Ceolotto et al[171], 2017 estern diet (WD);/- mice fed a W inhibitor achieves the reverse Inhibitor promotes RCT in vivo,elevates HDL levels, reduces aortic lipid deposition and plaque area in apoE-/- mice (WD)Mimic reduces hepatic expression of ABCA1 and high-density lipoprotein(HDL) levels, impairs reverse cholesterol transport and promotes atherogenesis in apoE-/- mice-Most abundant miR in murine macrophages; levels elevated in aortic plaque macrophages isolated from LDL receptor knockout (Ldlr-/-) (WD)mice;knockout of miR-21 enhances arterial macrophage accumulation,production of inflammatory cytokines Plasma levels correlate with plaque progression and vulnerability in patient with acute ischemic stroke.Long-term systemic delivery of antagomir reduces atheroma and promotes plaque stability (apoE-/-mice)Hsa-miR-24 levels inversely correlate with MMP-14 protein, and lesion instability Inhibitor increases lesion size and MMP-14 levels in apoE-/-mice (HFD)--Low serum levels of hsa-miR-30c-5p predict carotid atherosclerosis.Antagomir impairs endothelial Inhibitor enhances ABCA1 cholesterol efflux Mimic decreases cholesterol efflux to apoA-I, and increases macrophage cholesterol content LPS stimulation of miR-21 inhibits foam cell formation and reduces secretion of IL-6, IL-12, TNF-α MiR21-/- macrophages exhibit increased ABCG1 degradation and decreased cholesterol efflux, enhancing foam cell formation.Inhibitor enhances cholesterol efflux and decreases foam cell formation via upregulation of ABCA1/G1 expression Inhibitor increases macrophage invasive capacity Mimic decreases cholesterol efflux, and increases free cholesterol content in macrophages, but blocks uptake of oxLDL [Lipoprotein lipase (LPL), cluster of differentiation (CD36)] and inhibits cholesterol esterification-Cluster differentiation (CD) 36-dependent uptake of oxLDL reduces miR-30c-5p, enhancing IL secretion ABCA1 ABCA1 Toll like receptor 4 (TLR4); NF-κB Mitogen-activated protein kinase kinase 3(MKK3)ABCA1/G1 Matrix metallo-proteinase (MMP)-14 ABCA1 LDL receptor class A domain containing 3(LRAD3)Caspase 3 AcLDL + diosgenin oxLDL OxLDL ± LPS(lipopoly-saccharide)AcLDL oxLDL Colony stimulating factor (CSF)AcLDL oxLDL oxLDL Human: THP-1 macrophages;Murine:Peritoneal macrophages Human: THP-1 macrophages;264.7 macrophages 264.7 macrophages 264.7 macrophages 264.7 macrophages 264.7 macrophages Murine:RAW Murine:RAW Murine:Bone-marrow derived macrophages Murine:RAW Primary: Human monocytederived macrophages Human: THP-1 macrophages;Murine:RAW Murine:RAW Human: THP-1 macrophages miR-19b-3p (↓)miR-20a/b (-5p)miR-21-5p miR-21-5p miR-23a-5p (↑) miR-23a-3p miR-24-3p miR-27a/b (-3p)miR-28a-5p (↑)miR-30c-5p

Li et al[170], 2018 Zhao et al[172], 2014 Gao et al[173], 2018 Karunakaran et al[154],2015 Ouimet et al[153], 2017 Kim et al[174], 2017 Zhao et al[175], 2017 Dai et al[176], 2018 Hueso et al[177], 2016 Chen et al[178], 2018 Peng et al[179], 2016 healing following carotid injury(C57BL/6J mice)---Antagomir reduces atheroma in apoE-/- mice (WD)Inhibition restores defective autophagy in aorta and macrophages of Ldlr-/- mice Levels are elevated in individuals at risk of atherosclerosis Levels of miR-34a decrease, and expression of HDAC1 increases, in the aorta of apoE-/- mice fed a high methionine diet Mimic decreases expression of LOX-1 and lipid accumulation in the aortic root in apoE-/- mice (HFD);inhibitor achieves the reverse miR-125b levels are increased in atherosclerosis; siRNA-CD40 apoE-/-mice exhibit reductions in lesion area---Inhibitor promotes cholesterol efflux compared with C.pneumoniae control Vaspin decreases expression of miR-33a via inhibition of NF-κB, enhancing cholesterol efflux Inhibitor enhances mitochondrial respiration, and cholesterol efflux to apoA-I Mimic inhibits the breakdown of lipid droplets by repressing effectors of macrophage autophagy.Silencing promotes lipid droplet catabolism,aiding ABCA1-dependent cholesterol efflux Mimic decreases the expression of ABCA1 and promotes lipid accumulation in macrophages, while the inhibitor achieves the reverse Overexpression of miR-34a reduces HDAC1 levels in foam cells, while knockdown achieves the reverse;HDAC1 induces homocysteine (Hcy)dependent foam cell formation Mimic reduces expression of LOX-1 and inhibits foam cell formation;inhibitor achieves the reverse Silencing of CD40 downregulates levels of miR-125;LPS stimulates miR-125b expression Mimic reverses the pro-atherogenic impact of long non-coding (lnc)RNA NEAT1 on foam cell formation Mimic prevents TR-4 mediated Oxidized LDL (lectin-like) receptor 1 (LOX-1)ABCA1 ABCA1 Peroxisome proliferator activator receptor coactivator 1 (PGC1A), pyruvate dehydrogenase kinase 4 (PDK4), solute carrier family 25 member 25 (SLC25A25),nuclear respiratory factor 1 (NRF1),transcription factor A, mitochondrial(TFAM)Autophagy protein 5 (Atg5); autophagyrelated 12 (Atg12), microtubule-associated protein light chain 3 (Map11c3b), AMPactivated protein kinase κ1 (Prkaa1),lysosomal associated membrane protein 1(Lamp1), transcription factor EB (TFEB),Forkhead box O-3 (FOXO3)ABCA1 Histone deacetylase 1 (HDAC1)Oxidized LDL (lectin-like) receptor 1 (LOX-1)NF-κB-Testicular orphan nuclear receptor 4 (TR4)oxLDL oxLDL ± C.pneumoniae Vaspin ± LPS-AcLDL oxLDL oxLDL + Hcy oxLDL LPS oxLDL oxLDL 264.7 macrophages 264.7 macrophages 264.7 macrophages 264.7 macrophages Murine:RAW Human: THP-1 macrophages Human: THP-1 macrophages Human: THP-1 macrophages;Murine:Peritoneal macrophages Murine:Peritoneal macrophages Human: THP-1 macrophages Human: THP-1 macrophages Murine:Peritoneal macrophages Murine:RAW Murine:RAW Murine:RAW miR-30c-1-3p (↑)miR-33a/b (-5p) (↑)miR-33a-5p miR-33 miR-33 miR-33a-5p (↑)miR-34a-5p (↓)miR-98-5p (↓)miR-125b-5p (↑)miR-128- (↓)miR-133a

Lan et al[180], 2016 Ye et al[181], 2018 Ramirez et al[182], 2013 Hu et al[183], 2014 Li et al[184], 2015 Lin et al[185], 2017 Yang et al[186], 2017 Li et al[187], 2016 Chen et al[178], 2009 Tian et al[188], 2014-Mimic increases atherosclerotic lesions, release of proinflammatory cytokines and peritoneal macrophage lipid accumulation in apoE-/-(HFD)mice;inhibitor achieves the reverse Mimic reduces HDL levels in vivo(C57BL/6); inhibitor achieves the reverse Agomir inhibits RCT in vivo, and accelerates atherosclerosis in apoE-/-mice (HFD).Circulating levels of miR-144-3p correlate with acute myocardial infarction miR-146a mimic inhibits inflammation and plaque development in apoE-/- x Ldlr-/- and Ldlr-/- mice (HFD)Levels are elevated in foam cells, and clinical specimens from patients with atherosclerosis Aortic levels increased in hyperhomo-cysteinaemic apoE-/-mice--Antagomir decreases lipid accumulation in macrophages and lesion formation in apoE-/- mice(HFD). Level is up-regulated in CD14+ monocytes from coronary enhancement of lipid uptake via CD36 LPL activity and protein, inflammatory cytokines and cholesterol mass enhanced by miR-134 mimic;inverse achieved using an inhibitor-Mimic reduces cholesterol efflux to apoA-I in macrophages Mimic reduces cholesterol efflux and enhances expression of cytokines (IL-1,TNF-α, IL-6)Increases in miR-146a inhibit proinflammatory responses in macrophages(TNF-α)Inhibition promotes inflammation and lipid uptake during formation of foam cells Viral overexpression reduces expression of DNMT1, increases levels of adipocyte differentiation related protein (ADRP)and enhances cholesterol accumulation in foam cells;down-regulation achieves the reverse Mimic inhibits lipid accumulation,increasing cholesterol efflux to apoA-I and HDL;an inhibitor achieves the reverse. Down-regulation of ADIPOR2 replicates the impact of the miR-150 mimic-Enhances lipid uptake and reactive oxygen species production by macrophages Angiopoietin (ANGTPL)/lipoprotein lipase (LPL)ANGTPL4/LPL ABCA1 ABCA1 NF-κB TNF receptor (TNFR) associated factor 6(TRAF6)DNA methyl-transferase 1 (DNMT1)Adiponectin receptor 2 (ADIPOR2)-HMG-box transcription factor 1 (HBP1)oxLDL-Liver X receptor ligand T0901317 oxLDL oxLDL-oxLDL oxLDL oxLDL Human: THP-1 macrophages 264.7 macrophages-Human: THP-1 macrophages;Murine:Peritoneal and J774.1 macrophages Human: THP-1 macrophages Murine:Peritoneal macrophages(wild type and apoE-/-)Human: THP-1 macrophages-Human: THP-1 macrophages Human: Peripheral blood monocytes Murine:RAW miR-134-5p miR-134-5p miR-144-3p miR-144-3p miR-146a-5p miR-146b-5p (↑)miR-148a-5p miR-152-3p miR-150-5p (↑)miR-155-5p (↑)miR-155-5p (↑)
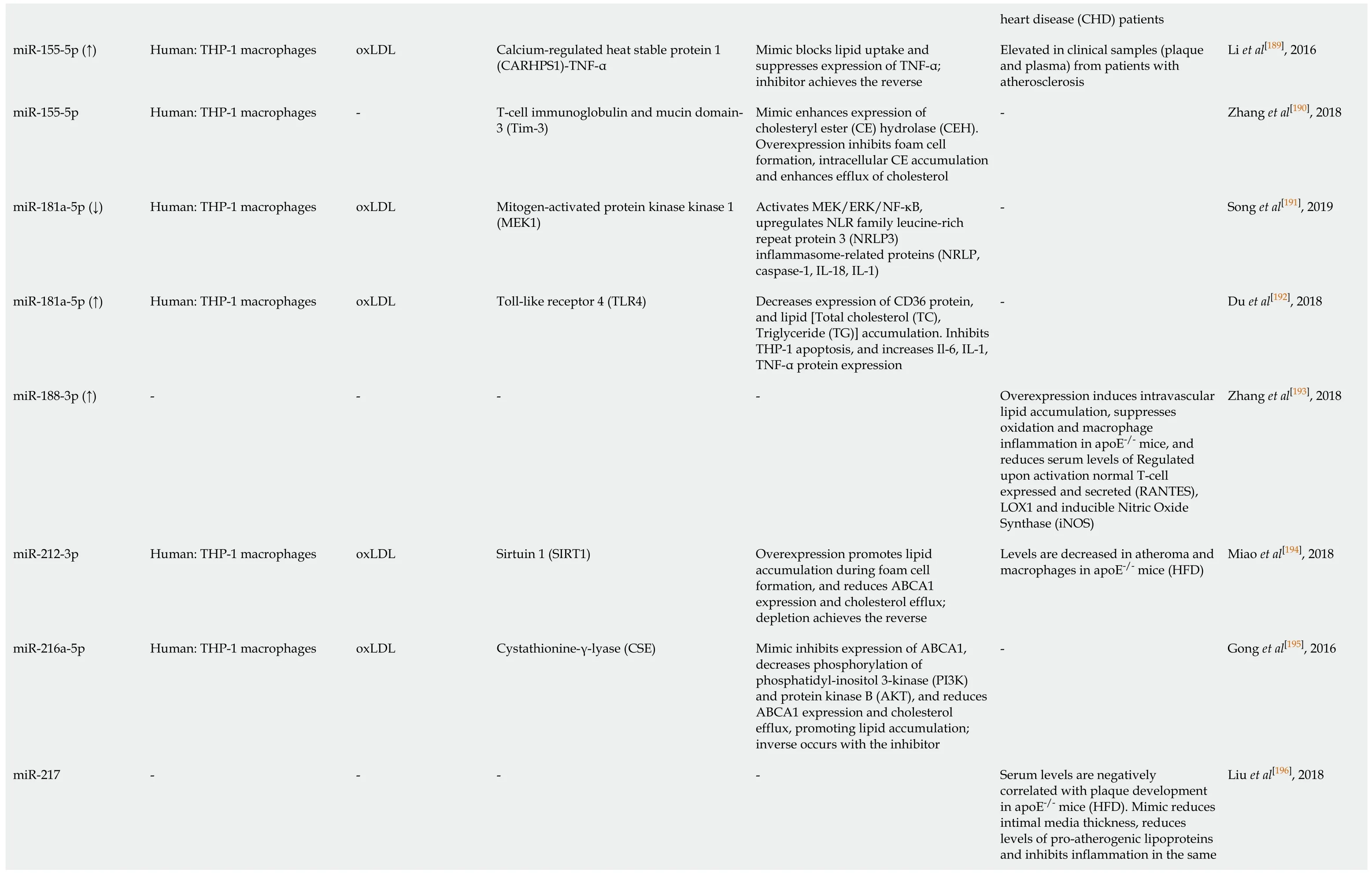
Li et al[189], 2016 Zhang et al[190], 2018 Song et al[191], 2019 Du et al[192], 2018 Zhang et al[193], 2018 Miao et al[194], 2018 Gong et al[195], 2016 Liu et al[196], 2018 heart disease (CHD) patients Elevated in clinical samples (plaque and plasma) from patients with atherosclerosis---Overexpression induces intravascular lipid accumulation, suppresses oxidation and macrophage inflammation in apoE-/- mice, and reduces serum levels of Regulated upon activation normal T-cell expressed and secreted (RANTES),LOX1 and inducible Nitric Oxide Synthase (iNOS)Levels are decreased in atheroma and macrophages in apoE-/- mice (HFD)-Serum levels are negatively correlated with plaque development in apoE-/- mice (HFD).Mimic reduces intimal media thickness, reduces levels of pro-atherogenic lipoproteins and inhibits inflammation in the same Mimic blocks lipid uptake and suppresses expression of TNF-α;inhibitor achieves the reverse Mimic enhances expression of cholesteryl ester (CE) hydrolase (CEH).Overexpression inhibits foam cell formation, intracellular CE accumulation and enhances efflux of cholesterol Activates MEK/ERK/NF-κB,upregulates NLR family leucine-rich repeat protein 3 (NRLP3)inflammasome-related proteins (NRLP,caspase-1, IL-18, IL-1)Triglyceride (TG)] accumulation. Inhibits THP-1 apoptosis, and increases Il-6, IL-1,Decreases expression of CD36 protein,and lipid [Total cholesterol (TC),TNF-α protein expression-Overexpression promotes lipid accumulation during foam cell formation, and reduces ABCA1 expression and cholesterol efflux;depletion achieves the reverse Mimic inhibits expression of ABCA1,decreases phosphorylation of phosphatidyl-inositol 3-kinase (PI3K)and protein kinase B (AKT), and reduces ABCA1 expression and cholesterol efflux, promoting lipid accumulation;inverse occurs with the inhibitor-Calcium-regulated heat stable protein 1(CARHPS1)-TNF-α T-cell immunoglobulin and mucin domain-3 (Tim-3)Mitogen-activated protein kinase kinase 1(MEK1)Toll-like receptor 4 (TLR4)-Sirtuin 1 (SIRT1)Cystathionine-γ-lyase (CSE)-oxLDL-oxLDL oxLDL-oxLDL oxLDL-Human: THP-1 macrophages Human: THP-1 macrophages Human: THP-1 macrophages Human: THP-1 macrophages-Human: THP-1 macrophages Human: THP-1 macrophages-miR-155-5p (↑)miR-155-5p miR-181a-5p (↓)miR-181a-5p (↑)miR-188-3p (↑)miR-212-3p miR-216a-5p miR-217

TargetScan and miRDB were used to confirm mouse miRNA target prediction in humans;where (↑↓) is not indicated, the level of miRNA was not confirmed altered by macrophage lipid accumulation.ABCA1:ATP binding cassette transporter A1;ABCG1:ATP binding cassette transporter G1;oxLDL:Oxidized low density lipoprotein;AcLDL:Acetylated low density lipoprotein;ADAM22:A disintegrin and metalloprotease-22;ADIPOR2:Adiponectin receptor 2);ADRP:Adipocyte differentiation related protein;AKT:Protein kinase B;ANGTPTL4:Angiopoetin-like 4;ApoE:Apolipoprotein E;ATG5:Autophagy protein 5;ATG12:Autophagy-related 12;CARHPS1:Calcium-regulated heat stable protein 1;CD:Cluster of differentiation;CEH:Cholesteryl ester hydrolase 1;CSE:Cystathionine-γ-lyase;CSF:Colony stimulating factors;DNMT1:DNA methyltransferase 1;ERK:extracellular signal-regulated kinase;FOXO3:Forkhead protein O3;HAT1:Histone acetyltransferase 1;HBP-1:HMG-Box transcription factor 1;Hcy:Homocysteine;HDAC1:Histone deacetylase 1;HFD:High fat diet;IL:Interleukin;ISG15:Interferon-stimulated gene 15;IKKα/β:Inhibitor kappa B kinase alpha/beta;iNOS:Inducible nitric oxide synthase;Lamp1:Lysosomal associated membrane protein 1;lncRNA:Long noncoding RNA;LPL:Lipoprotein lipase;LPS:Lipopolysaccharide;LRAD3:Low density lipoprotein receptor class A domain containing 3;Map11c3b:LC3 microtubule-associated protein light chain 3;MEK1:Mitogen activated protein kinase kinase 1;MEKK1:Mitogen-activated protein kinase kinase kinase 1;MKK3:Mitogen activated protein kinase kinase 3;MMP-14:Matrix metalloproteinase-14;NF-κB:Nuclear factor kappa B;NFAM:Transcription factor A, mitochondrial;NFIA:Nuclear factor 1A;NLRP3:NLR-family, leucine-rich repeat protein 3;NRF1:Nuclear respiratory factor 1;OLR1/LOX1:Oxidized low density lipoprotein (lectin-like) receptor 1;PDCD4:Programmed cell death 4;PDK4:Pyruvate dehydrogenase kinase 4;PGC1-γ:Peroxisome proliferator activated receptor-γ coactivator 1;PI3K:Phosphatidylinositol 3-kinase;Prkaa1:AMP-activated protein kinase-α1;RANTES:Regulated upon activation normal T cell expressed and secreted;SIRT1:Sirtuin 1, NAD-dependent protein deacetylase sirtuin 1;SLC25A25:Solute carrier family 25, member 25;SREBF:Sterol regulatory element-binding transcription factor;STK11:Serine/threonine kinase 11;TC:Total cholesterol;TFEB:Transcription factor EB;TG:Triglyceride;TLR4:Toll-like receptor 4;Tim-3:T-cell immunoglobulin and mucin domain-3;TNF-α:Tumour necrosis factor alpha;TR4:Testicular orphan nuclear receptor 4;TRAF6:TNF receptor (TNFR) associated factor 6;Vaspin:Visceral adipose tissue-derived serine protease inhibitor;WD:Western diet.
Multiple miRNA sequences target genes involved in macrophage cholesterol homeostasis
It is well established that miR-33, encoded by an intronic sequence withinSREBF2,plays a role in modulating cholesterol metabolism, in part by repressing expression of ABCA1[152].However, this sequence also represses effectors of macrophage autophagy[153]thereby inhibiting the breakdown of lipid droplets, and targets genes central to mitochondrial respiration, which are needed for effective cholesterol efflux to apoA-I[154].The expressions of ABCA1 and/or ABCG1 within the cholesterol efflux pathway are also targeted by miR-19b[162], miR-20a/b[164], miR-23a-5p[167], miR-27a/b[169],miR-144[182]and miR-378[201](Table 1), highlighting the complexity of the epigenetic regulation mediated by microRNA sequences.Equally, proteins involved in uptake of modified LDL are also modulated by miRNA sequences:TLR-4 by miR-21[165], miR-181a[192]and miR-223[198], LOX-1 by miR-30[170]and miR-98[176], CD36 by miR-181a[192]and miR-758[204], while LPL is targeted by miR-134[180,181]and miR-361[200](Figure 2).Storage of cholesterol as droplets of cholesteryl ester is modified by miR-9 targeting ofSOAT1[157], while cholesterol removal by autophagy is reduced by miR-17-5p dependent repression of Beclin-1[160].
Notably, a mimic of miR-134, which enhances LPL activity and protein expression and increases macrophage cholesterol mass, also promotes the production of inflammatory cytokines[180]and increases atheroma formation in the apoE-/-murine model of atheroma[181].Sequences repressing ABCA1 (miR-144[182], miR-302[199]) also enhance cytokine expression;a mimic of miR-144[183]accelerates lesion developmentin vivo, and circulating levels of this sequence correlate with acute myocardial infarction[183], while an inhibitor of miR-302 increases aortic and hepatic expression of ABCA1 and reduces plaque size and inflammation in Ldlr-/-mice fed a high fat diet[199].
miRNA sequences linking inflammation with cholesterol accumulation in macrophages

Figure 2 Key pathways involved in foam cell formation regulated by microRNA.
miRNA sequences which target the expression of proteins within cell signalling pathways mediating inflammatory responses have also been shown to reduce cholesterol accumulation in macrophages (Table 1)[153,155-204].For instance, let-7g inhibits both canonical (RelA/p50) and non-canonical (RelB/p52) NF-κB signalling pathways,limiting inflammatory (IL-1, IL-6, MCP-1) and apoptotic responses, and decreasing macrophage foam cell formation[155]in vitroandin vivo.Further, let-7g inhibition of nuclear translocation of RelA/p50 in macrophages treated with OxLDL prevents NFκB dependent upregulation ofSREBF2and miR-33a, and results in up-regulation of ABCA1[155].Indeed, aberrant expression of members of the lethal-7 (let-7) miRNA family have been linked with a number of diseases, including atherosclerosis and cancer[205-207]:Reductions in expression of let-7, which can be mediated by RNA binding protein Lin-28 homolog A (Lin-28), is observed in human carotid plaques from diabetic individuals, and diabetic apoE-/-mice[207].
miR-146a, which also targets the NF-κB pathway, inhibits the production of TNF-α by macrophagesin vitro, and limits inflammation and plaque development in murine models of atheroma[184].Plaque development and inflammation are also inhibited by miR-146a which targets the tumour necrosis factor receptor-associated factor (TRAF6)-NF-κB signalling axis[185]thought to underlie many cardiovascular pathologies[208].Equally, the loss of miR-21, which targets mitogen-activated protein (MAP) kinase kinase 3 (MKK3) within the p38 MAP kinase pathway[166], promotes the degradation of ABCG1, reducing cholesterol removal and promoting the formation of foam cellsin vitro.In vivo, deletion of miR-21 increases the number of macrophages within arterial lesions, and enhances the production of inflammatory cytokines[166].Reductions in expression of miR-181a, which targets mitogen-activated protein kinase kinase 1(MEK1) in the extracellular signal-regulated kinase (ERK)-1/2 pathway, have been linked to upregulation of NRLP3 inflammasome-related proteins[191], while increased expression of this sequence is associated with decreases in macrophage lipid accumulation[192].
Unsurprisingly, given their roles in regulating inflammatory responses, a number of miRNA sequences have been linked with regulating macrophage polarisation to differing phenotypes, recently reviewed by Essandohet al[209].Notably, miR-9, miR-125b and miR-155 are sequences linked with polarization towards the M1 phenotype[209];miR-125b and miR-155 are induced by exposure to oxLDL in human macrophages, but mimics of miR-9 and miR-155 are linked with inhibition of foam cell formation by repression ofSOAT1[157], enhanced expression of cholesteryl ester hydrolase[190], blockade of lipid uptake[189]and increased cholesterol efflux[190],suggesting divergence from the inflammation-lipid accumulation axis.Macrophages are induced to the M2 phenotype by several sequences, including miR-146a and miR-223[209].miRNA-146a inhibits inflammatory responses in murine macrophages, and also reduces inflammation and plaque formation in murine models of atheroma[184].Levels of miR-223 are reduced by OxLDL, and LPS, but elevated in murine atherosclerotic lesions, and overexpression of this sequence prevents both foam cell formation and production of inflammatory cytokines[198].However, much less is known about the impact of miRNA mimics or inhibitors involved in phenotypic modulation after induction of lipid accumulation in macrophages, and whether these molecules can induce phenotypic plasticity or aid lesion regression remains a key question.
Novel and emerging pathways associated with foam cell formation
Importantly, research into microRNA sequences modulated during foam cell formation has highlighted a number of previously unrecognised pathways contributing to this process, which may also prove useful therapeutic targets(Figure 3).For example, the study of miR-155 revealed a previously unsuspected role for calcium-regulated heat stable protein 1 (CARHSP1/CRHSP-24) in foam cell formation[189].This cytoplasmic protein, a cold shock domain (CSD) protein family member, is found within processing bodies or exosome granules, and was first identified as the physiological substrate for calcineurin (PP2B)[189,210,211].The conserved CSD domain binds to the AU-rich element (ARE) in the 3-UTR of TNF-α, increasing mRNA stability and enhancing inflammation[210,211].NF-κB induction of miR-155 by oxLDL in human macrophages is mirrored by increased levels in plasma and atherosclerotic lesions of patients with atherosclerosis[189].MicroRNA-155 binds directly to the 3’-UTR of CARHSP1 to reduce expression of this protein and TNF-α in macrophage foam cells;knockdown of CARHSP1 inhibits lipid accumulation and TNF-α production, while overexpression of CARHSP1 reverses the protective effects of miR-155[189].
Equally, insight into the hitherto uncharacterised role of programmed cell death 4(PDC4) in foam cell formation and atherosclerosis was revealed by investigation of the function of miR-16[159].Expression of PDCD4, which can act as a tumour suppressor, is induced by apoptosis and is known to regulate both inflammatory and apoptotic responses[212-214].MicroRNA-16 suppresses the activation of inflammatory macrophages by directly targeting the 3’-UTR of PDCD4[159].Levels of miR-16 decline in macrophages treated with oxLDL and in aortic lesions of apoE-/-mice fed a high fat diet, which also exhibit greater levels of PDCD4 protein.Either knockdown of PDCD4,or transfection with a miR-16 mimic, inhibits the expression and secretion of proinflammatory cytokines, and enhances expression and release of the anti-inflammatory factor IL-10, while an inhibitor of miR-16 achieves the reverse;these outcomes are also associated with modulation of ERK, p38 MAP kinase and NF-κB[212].
In other studies, the mechanism of action of molecules such as puerarin, the major bioactive ingredient isolated fromPueraria lobataand known as Gegen in traditional Chinese medicine, have been revealed by studies using miRNA[156].Targeting the 3-UTR region of serine/threonine kinase 11 (STK11) using a miR-7 mimic, revealed that this drug enhances ABCA1-dependent cholesterol effluxviaa mechanism which involves STK11 activation of AMP kinase and enhanced expression of PPAR-γ-LXRABCA1.Finally, it is clear that the contribution of some miRNA targets in foam cells remain to be established.For example, miR-28-5p, which is upregulated in murine macrophages treated with oxLDL, targets LDL receptor class A domain containing 3 (LRAD3)[170], but the contribution of this novel lipoprotein receptor to foam cell formation has not been investigated:At present, this protein has been linked with amyloid precursor protein trafficking in neurons[215]and with activation of E3 ubiquitin-protein ligase Itchy homolog (Itch) and E3 ubiquitin-protein ligase NEDD4 that promote proteasomal degradation[216].

Figure 3 Identification of novel pathways associated with foam cell formation.
Pathways targeted by miRNA sequences altered in human macrophage “foam”cells:DIANA/KEGG predictive analysis
It is increasingly recognised that networks of miRNA sequences, and their combined effects on multiple pathways, are important epigenetic determinants of complex phenotypes, just as genome-wide association studies have revealed shared genes and pathways in human disease[217,218].The (human) sequences described in Table 1 were analysed using DIANA-miRPATH v3.0, and the miRNA versus GO/GOSlim/KEGG entries heat map is shown in Figure 4.This functionality enables identification of miRNA belonging to similar functional categories, and identification of pathways lying under the regulation of similar miRNAs[219].
Several well-established pathways, targeted by multiple and distinct miRNA sequences/clusters, and known to regulate vascular function and atherogenesis,emerge from this predictive analysis.These include adherens junctions, which are a key part of the common signalling network linking age-related disease proteins(ARDPs) and longevity-associated proteins (LAPs) in the human interactome[220].The endothelial adherens junction complex, formed of vascular endothelial (VE)-cadherin and associated catenins, is a key determinant of arterial permeability, dysregulation contributing to vascular inflammation and atherosclerosis[221], while attenuating intraplaque vascular leakage reduces macrophage accumulation, necrotic core size and intraplaque haemorrhage[222].Identification of the TGF-β signalling pathway as a target of miRNA sequences altered in macrophage foam cells is equally unsurprising, given the widely recognised, and extensively reviewed, role of this cytokine in controlling macrophage phenotype[223], atherosclerosis[224]and cardiovascular function[225].

Figure 4 Pathways targeted by miRNA sequences altered in human macrophage “foam” cells:DIANA/KEGG predictive analysis.
More intriguingly, the Hippo signalling pathway emerges as highly targeted by multiple miRNA sequences implicated in foam cell formation (Figure 4);originally discovered inDrosophilaand highly conserved in mammalian cells, this pathway regulates cell survival, proliferation and apoptosis[226].Liet al[170]first showed that target genes of differentially expressed miRNA sequences are enriched in this pathway, in murine RAW264.7 macrophages treated with OxLDL;the Hippo/Yesassociated protein (YAP) signalling pathway is linked with vascular remodelling,pulmonary hypertension, aortic aneurysm, restenosis and angiogenesis, and atherosclerosis[226,227].Notably, the atheroprotective effect of steady laminar flow in major arteries is linked with inhibition of Hippo/YAP effector function[228]while activation of this pathway is linked with vascular remodelling, and switching of arterial smooth muscle cells to the “synthetic” proliferative phenotype in response to biochemical stretch[229].The effector function of YAP is linked with accelerated atherosclerosis in apoE-/-mice[230], while the herbal extractScutellarincan protect against atherosclerosis in rats by modulating the Hippo-YAP-Forkhead box (FOXO)3A transduction pathway[231].
Another pathway enriched in targets of multiple miRNA sequences is that involved in bacterial invasion of epithelial cells (Figure 4).Infection and systemic inflammation are linked with atherogenesis in a number of epidemiological studies[232]and vascular cells and macrophages are subject to invasion by bacteriaviaa number of mechanisms,including evasion of autophagy and internalisationvialipid rafts.In turn, this has led to the notion of vascular tissue providing a “privileged niche” in which bacteria can persist in dormancy for extended periods of time before becoming activated in phagocytic cells, contributing to the chronic and unresolved inflammation which characterises atherosclerosis[232].The epigenetic miRNA profile found in macrophage“foam” cells which may modulate susceptibility to bacterial invasion may also suggest key proteins (and pathogens) implicated in this process, and/or highlight possible therapeutic strategies designed to limit the impact of vascular “infectology”[232].
THERAPEUTIC OPTIONS:CLINICAL APPLICATIONS OF MIRNA(TARGETS)
miRNA pathways are excellent candidates for pharmacological manipulation, and have been invoked as biomarkers, diagnostics or therapeutics for a number of disease conditions[140,233,234].For example, Carusoet al[233]monitored dynamic changes in microRNA profiles in lung tissue during the development of pulmonary arterial hypertension (PAH) in hypoxic rodents.The same authors discovered that miR-145 is a useful indicator of hypoxia in mice, and of heritable and idiopathic pulmonary arterial hypertension in patients;down-regulation of miR-145 also had utility in protecting against development of PAH in mice[234].Further, the “ThyraMIR” testing platform, which examines the expression of a panel of ten miRNA in conjunction with selected disease-associated genes, has been approved for diagnostic use in thyroid cancer when malignancy risk cannot be determined by conventional cytology[235].
Treatments involving miRNAs focus on the concept of specifically influencing levels of miRNAs in certain diseases – including suppression of miRNAs, as well as raising miRNA levels or substituting artificially generated copies[140].Mimics can be used for gene silencing, by generating artificial, double-stranded miRNA-like RNA fragments,which bind specifically to target mRNA, activating the RISC complex;this results in down-regulation of specific mRNAs and gene suppression (above).Equally,chemically engineered oligonucleotides are capable of silencing single endogenous miRNAs, binding to the target mature miRNA, leading to reduced activation of RISC and up-regulation of specific mRNAs and gene expression.Other approaches involve“target mimicry” using miRNA sponges, masking or erasers[140].
Delivery of disease-specific miRNA mimics or inhibitors remains in the developmental stage with multiple miRNA therapeutics currently in clinical trials.The most advanced trial, currently in Phase II, employs a chemically engineered inhibitor for miR-122 (Miravirsen) which, under normal conditions, binds to the 5’-UTR region of the hepatitis C virus and enhances its transcription[236-243].By hybridizing to mature miR-122, Miravirsen has been shown to effectively inhibit viral replication with minimal “off target” effects[236-239].MicroRNA-based clinical trials are also underway for the development of novel treatments for various cancers.Currently in Phase I and Phase II trials, the efficacy of an inhibitor (MRG-106) targeting miR-155 is being investigated for treatment of a variety of lymphomas, reflecting the recognised role of this sequence in driving malignant lymphocyte proliferation[240,241].
Despite the encouraging progression of miRNA therapies, significant challenges have also been highlighted in some clinical trials.One such promising miRNA therapeutic, the miR-34 mimic “MRX34”, was employed in a Phase I trial for patients with advanced liver cancer[242-244];despite dose-dependent modulation of miR-34 target oncogenes, the study was halted due to serious adverse effects in a small cohort of subjects[242-244].This study also highlighted a challenging area for the development of miRNA-based therapies:The preclinical studies demonstrated that the liposomal delivery system resulted in elevated miR-34 in multiple tissues in non-human primates[244].While this may be beneficial for miRNA therapeutics used to treat diseases that can arise in several anatomical locations, in the case of tissue specific diseases, such as atherosclerosis, site-specific homing could dramatically reduce potential off-target effects.
To overcome this issue, liposomes enriched in specific amino acid sequences have been developed which result in increased tissue-specific accumulation.The efficacy of this system, for the delivery of short, siRNA, has been demonstratedin vivoin osteoporotic mice[245].Use of a lipid nanoparticle containing C-C chemokine receptor type 2 (CCR2)-targeting siRNA resulted in high levels of localisation in bone marrow and spleen, significant reductions in monocyte CCR2 expression, decreased myeloid cell infiltration in the plaque and an overall reduction in lesion size in ApoE-/-mice[246].While this system targets atherosclerotic plaque indirectly, additional delivery mechanisms have been employed in animal studies that may facilitate plaque-directed delivery of miRNA-based therapeutics.Notably, reconstituted HDL (rHDL) can act as a carrier particle for delivery of drugs and microRNA:In ApoE-/-mice, rHDL was used to delivery simvastatin to plaque regions, resulting in reduced local inflammation[247],while miR-223 incorporated into rHDLin vitrowas able to selectively target cells expressing SR-BI[148].
Thus, many factors need careful consideration in developing miRNA therapeutics for atherosclerosis, including effective vectors and delivery options, and the nature of“off-target” side-effects and/or toxicities which may occur due to disruption of multiple target genes and/or cell signalling networks[248,249].However, since differing microRNA sequences impact on distinct stages of the atherogenic process[248,249],delivery of a pool of mimics and/or inhibitors may be an attractive therapeutic strategy for treatment of this complex, multicellular disease[248,249].Defined stages of the disease process could be targeted by distinct miRNA mimics/inhibitors, predicated by serum levels of secreted miRNA sequences.Such approaches, if fully validated, might be used to provide personalised treatment, or be beneficial in targeting asymptomatic patients, or those in whom statin use is contraindicated or ineffective[250-252].
CONCLUSION
Huge advances have been made in understanding the epigenetic factors, and particularly the role of small non-coding miRNA sequences, in regulating macrophage“foam” cell formation and function over the last decade.Networks of genes regulated by multiple miRNA sequences have been revealed, and new pathways discovered which contribute to the atherogenic process, which may ultimately lead to RNA-based therapeutics capable of preventing or regressing the formation of complex atherosclerotic lesions by targeting macrophage function.
