基于高通量芯片对小儿急性髓系白血病的生物信息学分析
2020-10-21张曦翟丽孙运艳杨伟高艳章雷鸣潘玉卿
张曦 ,翟丽 ,孙运艳,杨伟,高艳章,雷鸣,潘玉卿
(1.云南省肿瘤医院·昆明医科大学第三附属医院检验科,云南 昆明 650118;2.云南省肿瘤医院·昆明医科大学第三附属医院血液科,云南 昆明 650118;3.昆明医科大学第一附属医院检验科云南省实验诊断研究所云南省检验医学重点实验室,云南 昆明 650032)
小儿急性髓系白血病(acute myeloid leukemia,AML)约占儿童白血病总数的20%[1-2]。近年来,由于白血病诊疗方法和支持治疗的改进,AML 患儿的长期生存率有了很大提高。虽然小儿AML 的预后明显优于20 岁以上的成人[3-5],但由于其化疗耐药、缓解率低和缓解期感染等因素阻碍了治愈率的提高[6],尤其是不明原因发热、体质量减轻、肺部浸润伴呼吸困难和肾功能衰竭等并发症在治疗过程中时有发生[7]。即使在发达国家,对复发难治性小儿白血病的治疗效果仍不乐观[8]。目前国内外对于小儿AML 的治疗主要以放、化疗及造血干细胞移植为主,但对于其发病机制的报道相对较少,故探究小儿AML 的分子机制并制定精准的治疗策略对于患儿的生存及预后尤为重要。迄今为止,白血病的病因尚不明确,其仍然是儿童和青少年癌症死亡的主要原因。本研究利用生物信息学分析手段对小儿AML 患儿基因芯片进行分析,以期发现与该病相关的生物标志物,为揭示小儿AML 发生的分子机制提供理论依据。
1 资料与方法
1.1 小儿髓系白血病基因芯片数据信息获取从GEO 数据库(http://www.ncbi.nlm.nih.gov/geo)下载相关的基因表达矩阵,以“pediatric acute myeloid leukemia”为关键词筛选相关数据集。筛选条件:①mRNA 表达谱数据集;②以骨髓为样本;③以正常骨髓为对照。选择基于GPL8300平台的 GSE2191 数据集(GeneChip Human Genome U95Av2 oligonucleotide microarray),包括54 例小儿AML 患儿(男性33 例,女性21 例,年龄0 d~15岁)和4名正常骨髓样本(正常人)。此外,本研究不包括任何对人体细胞或动物进行的实验。
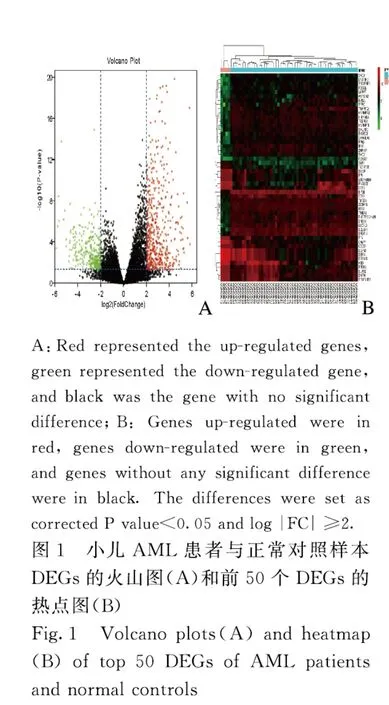
1.2 基因芯片的处理及差异表达基因(differentially expressed genes,DEGs)的筛选采用GEO 数据库自带的GEO2R 在线工具对AML 患儿及正常对照组间的DEGs 进行筛选[9],筛选条件:错误发现率(false discovery rate,FDR)<0.05 且|log2fold change(FC)| ≥2。以logFC ≥2 为上调基因,logFC ≤-2 为下调基因。下载MINiML 平台文件,采用R 语言的ggplot 包绘制DEGs 的火山图,pheatmap 包绘制差异较大的前50 个DEGs 的热点图。
1.3 DEGs 的基因本体功能注释(GO)富集和京都基因与基因组百科全书(KEGG)通路分析利用DAVID 6.8 数据库分析GO 富集和KEGG 通路富集情况[10]。GO 分析中以FDR <0.05、基因数(gene counts)>10 作为筛选条件。KEGG 分析中以P<0.05 作为筛选条件。采用R 语言的GOplot包对GO 富集实现可视化,并获得分子功能(molecular function,MF)、生物学过程(biological process,BP)和细胞成分(cellular component,CC)等注释分析。
1.4 蛋白互作网络(PPI)网络构建与核心基因(Hub gene)筛选采用 SRTING 数据库(https://stringdb.org)对本次研究中得到的DEGs 构建PPI 网络[11],并将得到的数据下载并导入Cytoscape 软件(www.cytoscape.org/)进行可视化并去除游离蛋白质节点后,行CytoHubba 插件计算每个节点的连接度(Degree),将连接度按照降序排列,并获得排名前20 位的Hub 基因[12]。
1.5 Hub 基因参与疾病谱及相关转录因子分析将前5 位的Hub 基因通过生物技术信息基因云(gene-cloud of biotechnology information,GCBI)在线数据库进行分析,并采用Cytoscape 中的iRegulon 插件对20 个Hub 基因的相关转录因子进行预测,筛选条件为标准化富集分数(normalized enrichment score,NES)>3[13]。
2 结果
2.1 AML 患儿DEGs 的筛选运用GEO2R 功能进行DEGs 识别,按照上述的DEGs 筛选条件得到600 个DEGs,其中407 个基因上调,193 个基因下调。数据集中的上调和下调基因如图1A所示。前50 个DEGs 如图1B所示。
2.2 DEGs 的GO 富集和KEGG 通路分析将DEGs 上传到DAVID 数据库,获得GO 功能富集和KEGG 通路分析。GO 分析结果显示:DEGs 主要富集在细胞成分中,包括核浆、细胞质、核膜和核斑点;在分子功能层面,DEGs 主要涉及蛋白质及RNA 的poly(A)尾结合;在生物学过程层面,DEGs 主要涉及白细胞迁移。见图2和表1。KEGG 通路富集结果显示:DEGs 主要富集在肿瘤坏死因子、细胞因子受体相互作用和Jak-STAT 信号通路中。见表2。
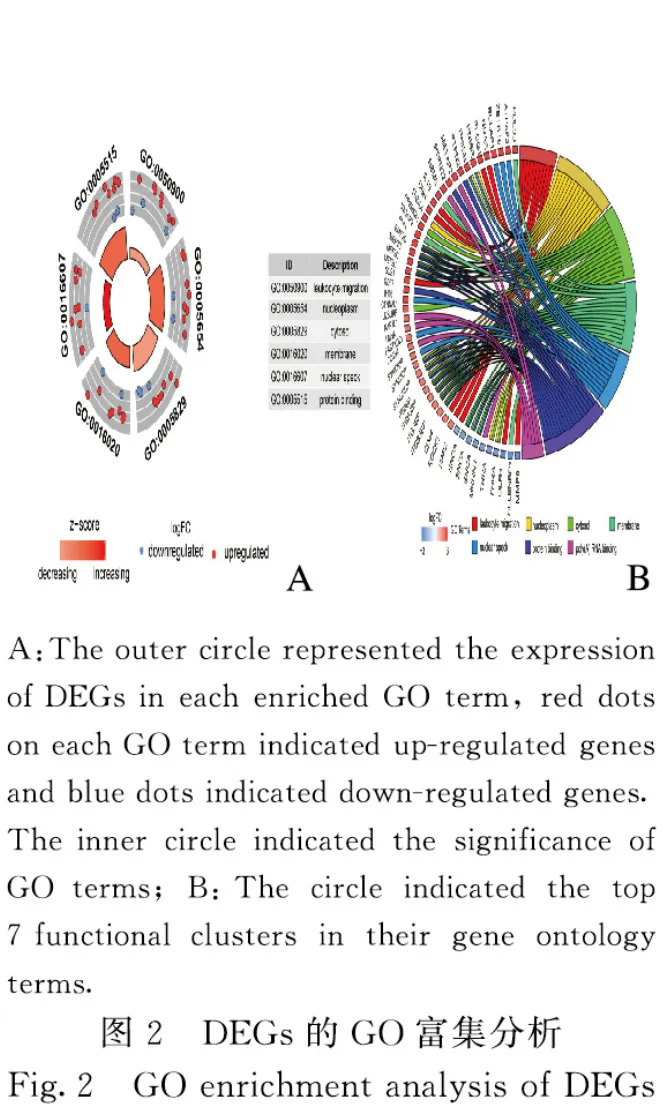
表1 DEGs 在GO 富集中的分布Tab.1 Distribution of DEGs in GO enrichment

表2 DEGs 的KEGG 通路富集分析Tab.2 KEGG pathway analysis of DEGs
2.3 PPI 网络构建与Hub 基因筛选利用STRING 数据库构建PPI 网络,网络共涉及551 个节点和773 个连接。计算每个蛋白质节点的连接度,结果表明:PPI 网络的最大连接度为24,最小连接度为16(图3)。排名前20 位的Hub 基因分别是甲酰肽受体2(formyl peptide receptor 2,FPR2)、磷酸肌醇3 激酶调节亚单位1(phosphoinositide-3-kinase regulatory subunit 1,PIK3R1)、E1A 结合蛋白p300(E1A binding protein p300,EP300)、热休克蛋白90α 家族AA1(heat shock protein 90 alpha family AA1,HSP90AA1)、NRAS 原癌基因(NRAS protooncogene,NRAS)、精氨酸酶1(arginase 1,ARG1)、磷脂酰肌醇4,5-二磷酸3-激酶催化亚单位 α(phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha,PIK3CA)、前血小板碱性蛋白(pro-platelet basic protein,PPBP)、CD59 分子(CD59 molecule,CD59)、细胞骨架相关蛋白4(cytoskeleton associated protein 4,CKAP4)、乙酰辅酶a 酰基转移酶1(acetyl-CoA acyltransferase 1,ACAA1)、抗菌肽(cathelicidin antimicrobial peptide,CAMP)、肽聚糖识别蛋白1(peptidoglycan recognition protein 1,PGLYRP1)、磷脂酶γ-1(phospholipase C gamma 1,PLCG1)、基质金属肽酶8(matrix metallopeptidase 8,MMP8)、乳糖转铁蛋白(lactotransferrin,LTF)、转氨酶1(transcobalamin 1,TCN1)、嗅介蛋白4(olfactomedin 4,OLFM4)、结合珠蛋白(haptoglobin,HP)和富半胱氨酸分泌蛋白3(cysteine rich secretory protein 3,CRISP3)。见表3。

2.4 参与白血病的Hub 基因和相关转录因子筛选GCBI 分析结果显示:EP300、HSP90AA1和NRAS 共3 个Hub 基因参与了小儿AML 的发生发展(图4);通过Cytoscape 中的iRegulon 插件预测DEGs 中排名前20 个Hub 基因的转录因子,结果显示:共有55 个转录因子调节DEGs,排名前15 位转录因子结果见表4。部分转录因子的作用关系见图5。

表3 连接度最高的排名前20 个Hub 基因Tab.3 Top 20 Hub genes with higher degree of connectivity
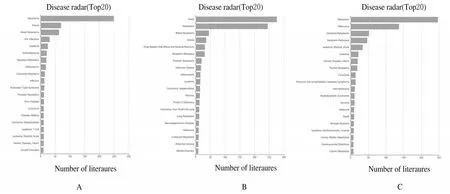
图4 Ep300(A)、HSP90AA1(B)和NRAS(C)的相关疾病谱Fig.4 Disease spectrum of EP300(A),HSP90AA1(B),and NRAS(C)

表4 与Hub 基因相关的排名前15 位转录因子(NES >3)Tab.4 Top 15 transcription factors associated with Hub genes (NES >3)
3 讨论
1~14 岁儿童所患肿瘤中近1/3 是白血病,其中 3/4 为急性淋巴细胞性白血病(acute lymphoblastic leukaemia,ALL)[14]。虽然小儿AML 所占比例不大,但如果不经任何治疗,AML会在发病初期对儿童造成极大的危害[15]。在白血病发生发展过程中参与造血调控的基因常发生突变,导致造血细胞分化缺陷。而每种类型的白血病都有不同的基因突变。临床上,白血病治疗和生存率主要取决于基因突变的类型和诊断分期[16]。为此,本研究从GEO 数据库下载小儿AML 的基因表达矩阵,筛选出可能参与小儿AML 的核心基因,旨在为小儿AML 的发病机制、诊断及防治提供新的理论依据和研究视角。
本研究采用STRING 数据库构建PPI 网络后,通过Cytoscape 软件对网络进行可视化。利用CytoHubba 插件计算每个蛋白质节点的连接度,共筛选出Ep300、HSP90AA1 和NRAS 等20 个与小儿AML 相关的Hub 基因,对上述基因进行文献检索后发现:对维甲酸处理的急性早幼粒细胞能够显著增加细胞分化时膜表面的抗炎因子Annexin A1及其受体分子FPR2 的表达[17]。对诱导化疗失败的AML 患儿进行全基因组DNA、转录组RNA 和miRNA 测序结果显示:PIK3R1 参与小儿AML 早期化疗耐药的遗传机制[18],而PIK3R1 作为白血病的超级增强子抑制了细胞凋亡并促进其增殖[19]。Ep300 基因表达下调并作为抑癌因子参与小儿AML 的进程[20]。在造血干细胞和祖细胞内,由于Ep300 基因的缺失直接增强了MAPK 及JAK/STAT 等细胞因子的信号通路,Ep300 基因甚至能够阻断造血异常综合征(human myelodysplastic syndrome,MDS)向AML 的转变[21]。HSP90AA 1 基因作为重要的分子伴侣参与细胞增殖、存活和适应的信号传导途径。抑制HSP90AA1 基因的表达活性后,能将其作为研究AML 的重要分子靶标[22]。单核苷酸变异分析[23]显示:NRAS 在初诊为急性早幼粒细胞白血病且维甲酸基因发生重排的患儿体内发生突变并参与了白血病的发病机制。NRAS 基因作为热点区域突变基因亚群,在小儿AML 的分子流行病学和生物学研究中具有重要意义[24]。通过维甲酸及1,25 二羟基维生素D3 的诱导,髓系白血病细胞能够成功向M2 型巨核细胞进行分化,此时其表面标记分子ARG1 的表达上调,提示其能够成为AML 化疗成功与否的标志物[25]。
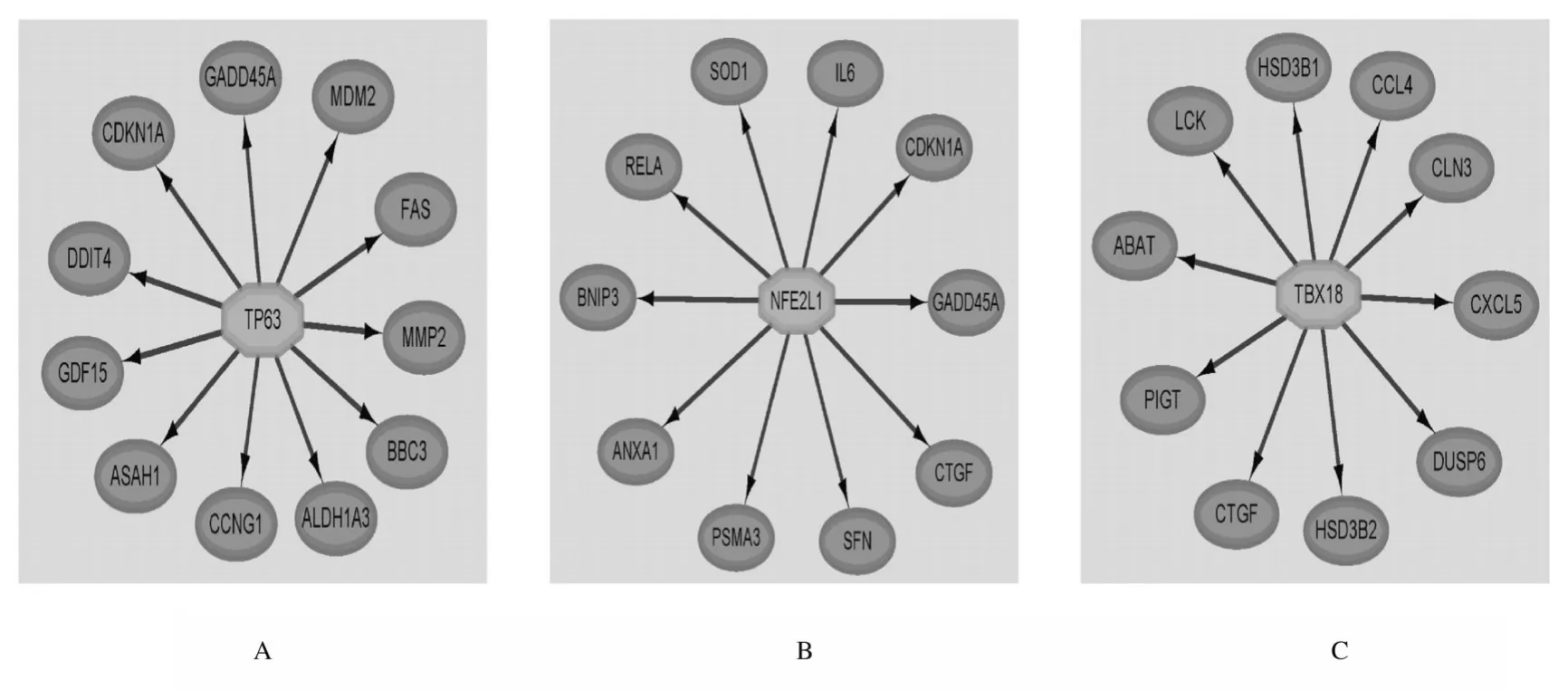
图5 转录因子的靶基因作用关系图Fig.5 Interaction networks of target genes for transcription factors
此外,本研究还应用Cytoscape 中的iRegulon插件预测Hub 基因的转录因子,并分析转录因子所调节的DEGs,结果显示:与20 个Hub 基因相关的转录因子有55 个,其中有部分转录因子在白血病中的作用已经得到证实。TP63 与长链非编码RNA rs55829688 发生相互作用,加剧了AML 患者的骨髓抑制,并影响该病的预后[26]。在多发性骨髓瘤中,长链非编码RNA MALAT1 的拮抗作用下调了蛋白酶体亚单位基因的2 个主要转录激活因子(NFE2L1 和NRF2),导致胰蛋白酶、糜蛋白酶和细胞凋亡蛋白酶活性降低,并导致多泛素蛋白的累积[27]。
综上所述,本研究采用GEO 数据库中的小儿AML 芯片信息,利用一系列的生物信息学手段,探寻小儿AML 相对精准的预后标志物,为其诊治提供更为有效的数据支持和理论依据。
