酸性橙II纯度标准物质的研制
2020-10-17周瑾艳黄彦捷许俊斌黄梓宸花秀兵王世超
周瑾艳,黄彦捷,许俊斌,黄梓宸,陈 玲,林 铿,花秀兵,王世超
(广东省计量科学研究院,广东 广州 510405)
1 引 言
食品成分分析是用化学方法对食品中含有的各种物质进行分析,包含食品内源成分、食品添加剂和污染物等的无机成分及有机成分进行分析。近年来,由滥用食品添加剂和非法添加非食用添加剂而引起的食品安全事件屡见不鲜,食品安全问题引起了世界各国政府的关注。食品成分分析标准物质作为一种可溯源性工具,广泛应用于食品添加剂市场的治理和规范,是保证分析结果正确和可靠的重要支撑[1]。然而,由于我国食品工业起步较晚,市面上适用的标准物质种类尚显不足,并且研发成本也不低于国外标准物质的销售价格。在食品安全问题日益成为社会焦点的今天,如何更好地促进食品及食品添加剂行业的健康有序发展,有效保障广大消费者的健康和权益,严格控制食品添加剂的过度使用,严厉打击食品中非法添加非食用物质的行为,是摆在我们面前的重大课题。我国已制定公布303部食品安全国家标准,覆盖6 000余项食品安全指标。这就要求国内的标准物质研发机构开发出一系列质量可靠、价格低廉的食品成分分析国家标准物质。
食品成分分析标准物质的研制过程通常包含标准物质样品的制备、均匀性检验、稳定性检验、定值、不确定度评定及比对和验证等几个程序[2,3]。每道程序均需要严格的实验过程、严谨的实验操作和严密的结果计算。在食品成分中,无机物成分包括水和无机质两种,有机物成分则包括色素成分、香气成分、呈味成分等。相对而言,分析食品成分中的有机成分的难度高于分析无机成分。有机成分的分析主要包括主成分定性分析和结构鉴定,纯度和含量测定[4]。有机物化学纯度的测定可以通过两种方法实现:(1) 直接测试主成分含量方法,主成分含量即为纯度值,主要通过色谱法、凝固点下降法、差示扫描量热法、定量核磁共振法、元素分析法、滴定法等手段实现;(2) 杂质扣除法即质量平衡法,测量所有可能的杂质的质量分数,从100%中扣除这些测得的杂质百分含量,即为纯度值[5]。有机杂质的测定多采用高效液相色谱法(high performance liquid chromatography,HPLC)[6],有时也采用薄层色谱法(thin layer chromatography,TLC)、气相色谱法(gas chromatography, GC)等方法。高效液相色谱-紫外检测器法(HPLC-UV/Vis)可实现对大多数有机物的检测,有些对紫外无响应的有机杂质则可采用通用性检测器如电雾检测器(CAD)等方法来实现。
本文选取的酸性橙Ⅱ是一种化工色素,工业上主要用于皮革、羊毛、纸张、蚕丝、锦纶等染色。食品工业中,酸性橙Ⅱ并不属于食用色素;因为人在食用酸性橙Ⅱ后会引起食物中毒,长期食用甚至会致癌,因此被明令禁止添加[7]。但一些不法商贩利用其色泽鲜艳、着色力强、价格低廉的特点,为求牟利非法生产与加工,从而严重危害消费者的身体健康。因此研制酸性橙Ⅱ纯度标准物质,用于食品行业相关分析方法的评价和相应仪器的校准就显得尤为重要和迫切。
本文将介绍利用经典的质量平衡法研制GBW(E)100373酸性橙Ⅱ纯度标准物质的过程,重点介绍研制过程中标准物质样品的制备、均匀性检验、稳定性检验、定值、不确定度评定及比对和验证。
2 质量平衡法
质量平衡法进行标准物质分析具有较高的准确度,被世界卫生组织(WHO)[8,9]、欧洲药典[10]推荐为药品标准物质定值方法,成为有机物纯度标准物质定值的常用手段。有机物纯度物质的主要杂质有与主成分结构类似的有机杂质、水分、残留溶剂和不挥发性杂质等[5]。测定过程中,质量平衡法克服了由于主成分和各类杂质理化性质不同,引起仪器响应值的差异,从而无法准确测定杂质含量的缺点。同时,该方法通过分析尽可能存在的杂质实现高准确度的纯度定值[11]。
2.1 与主成分结构类似的有机杂质分析方法
主成分及其结构类似的有机杂质可采用色谱法或其联用技术同时进行分析。色谱法分为液相色谱法和气相色谱法,及其后续不同联用技术法。通常采用高效液相色谱-紫外检测器法或二极管阵列检测器法(HPLC-UV/Vis或HPLC/PDA)针对包含紫外/可见吸收发色基团进行分析;采用气相色谱氢火焰法(GC/FID)分析挥发性和半挥发性的有机化合物。黄亮等[12]采用液相色谱-二极管阵列法分析了肌酐纯度标准物质,同时准确测试了水分、灰分、挥发性物质和无机元素含量,并采用定量核磁共振法对其定值进行比对[13,14];马康等[15]采用了气相色谱串联质谱(GC-MS)对1,2,3,4-四氯苯中的非挥发性杂质用进行定性,经标准添加的方式确认杂质为1,2,3,5-四氯苯,并用气相色谱电子捕获检测器(GC-ECD)对杂质进行准确定量;徐鹏等[16]深入研究并优化了高效液相色谱-二极管阵列检测器法(HPLC-DAD)与气相色谱-氢火焰离子化检测器法(GC-FID)对氯菊酯主成分的定值方法。
2.2 水分分析方法
有机物的水分分析对其纯度定值至关重要,由于大部分高纯有机物水分含量较低,因而对分析方法的灵敏度和精密度要求较高,常见的有卡尔费休法和热重法。其中热分析技术是通过加热使样品水分蒸发,用热重(thermogravimetric analysis,TG)曲线监测蒸发的水分重量。该方法简单直观,但要求样品在分析温度范围内中不发生分解;同时,有些不易逸出的结晶水和易挥发性溶剂,均可能影响水分含量的测定结果。Yazgan等[17]采用热重-质谱联用技术全面分析了热失重过程中逸出的水分、降解产物和易挥发性物质。
除了卡尔费休滴定之外,根据水测定原理,也可通过蒸馏将水与样品物理分离,利用色谱法、烤箱红外线、微波干燥等方法测定水分含量;但需要大型实验设备,且由于样品中常存在挥发性杂质,因此这些方法存在很大的局限性。由于水分含量会影响样品的物理性质,因而也可以通过光密度、极化、折射或电气方法来确定[18],但也不普遍适用。
2.3 挥发性有机物分析方法
有机物纯度物质在合成和提纯工艺中会存在溶剂残留,属于挥发性有机物。世界卫生组织(WHO)将挥发性有机物(VOCs)定义为熔点低于室温、沸点范围在50~260 ℃之间的挥发性有机化合物。检测VOCs常用的方法有气相色谱法和气相色谱-质谱联用、离子迁移谱和质子转移反应质谱法[20]。气相色谱是一种常用的VOCs分析方法,可用于痕量和痕量组分的定量。检测过程分为:收集气体样品、预处理、富集、气相色谱分离,然后使用合适的检测器(FID,MS等)进行检测。马康等[15]采用顶空GC-MS分析1,2,3,4-四氯苯中的溶剂残留,确定为乙醚,采用GC-FID对其准确定量。
2.4 不挥发性杂质分析方法
不挥发性杂质主要包括灰分和无机杂质。常用灼烧法和热重分析测定灰分,电感耦合等离子发射光谱(inductively coupled plasma,ICP)和电感耦合等离子体质谱(inductively coupled plasma mass spectrometry,ICP-MS)测定无机杂质。ICP-MS因其灵敏度优异、检测下限低、动态线性范围宽,成为目前痕量和超痕量无机元素的主要分析手段[21];通常采用热重分析和灼烧残留法测试灰分,作为验证。黄亮等[12]采用ICP-MS分析测定肌酐中73种无机元素的含量,总和为0.012%;同时用灰分热重法(TGA)进行灼烧,测定灰分总和为0.025%。
3 酸性橙Ⅱ标准物质样品的研制
3.1 标准物质候选物的提纯及分装
使用市售的高纯度酸性橙Ⅱ作为原料,用乙醇作为溶剂溶解,重结晶提纯,过滤晶体,并在105 ℃下干燥。过筛,混合并干燥后,置于装有五氧化二磷的干燥器中干燥24 h,备用。
样品经采用液相色谱-PAD检测法进行初步均匀性检验后,分装于5 mL的棕色样品瓶中,每瓶装200 mg,拧紧瓶盖后,用封口膜密封瓶口,置于低温(4 ℃)避光保存,一次分装400瓶。
3.2 标准物质样品的定性分析
对于有机分子,通常可以采用红外光谱、核磁共振、元素分析或是质谱的方式对其主要成分进行分析。其中,红外光谱是通过分子的特征官能团的伸缩振动和弯曲振动,观察其光谱中特定吸收/投射率的位置和强度,从而进行定性分析;而核磁共振常用1H谱和13C谱,通过不同原子的化学位移、偶合常数、积分面积等参数,推测分子中H原子或C原子所处的化学环境及相对数目;质谱主要通过寻找分子离子峰或特征碎片峰来定性分析相对分子质量;元素分析则可以实现有机成分中的C、H、O、S、P、F、Cl、Br等元素的准确定量。
图1给出了酸性橙Ⅱ纯度候选标准物质的红外光谱,可以从位于1 100~1 300 cm-1的芳环骨架振动,及1 500~1 750 cm-1的双键(N=N,S=O)振动进行定性分析,并通过和(spectral database for organic compounds,SDBS)数据库中的标准谱图比对,从而确定候选标准物质确实为酸性橙Ⅱ。
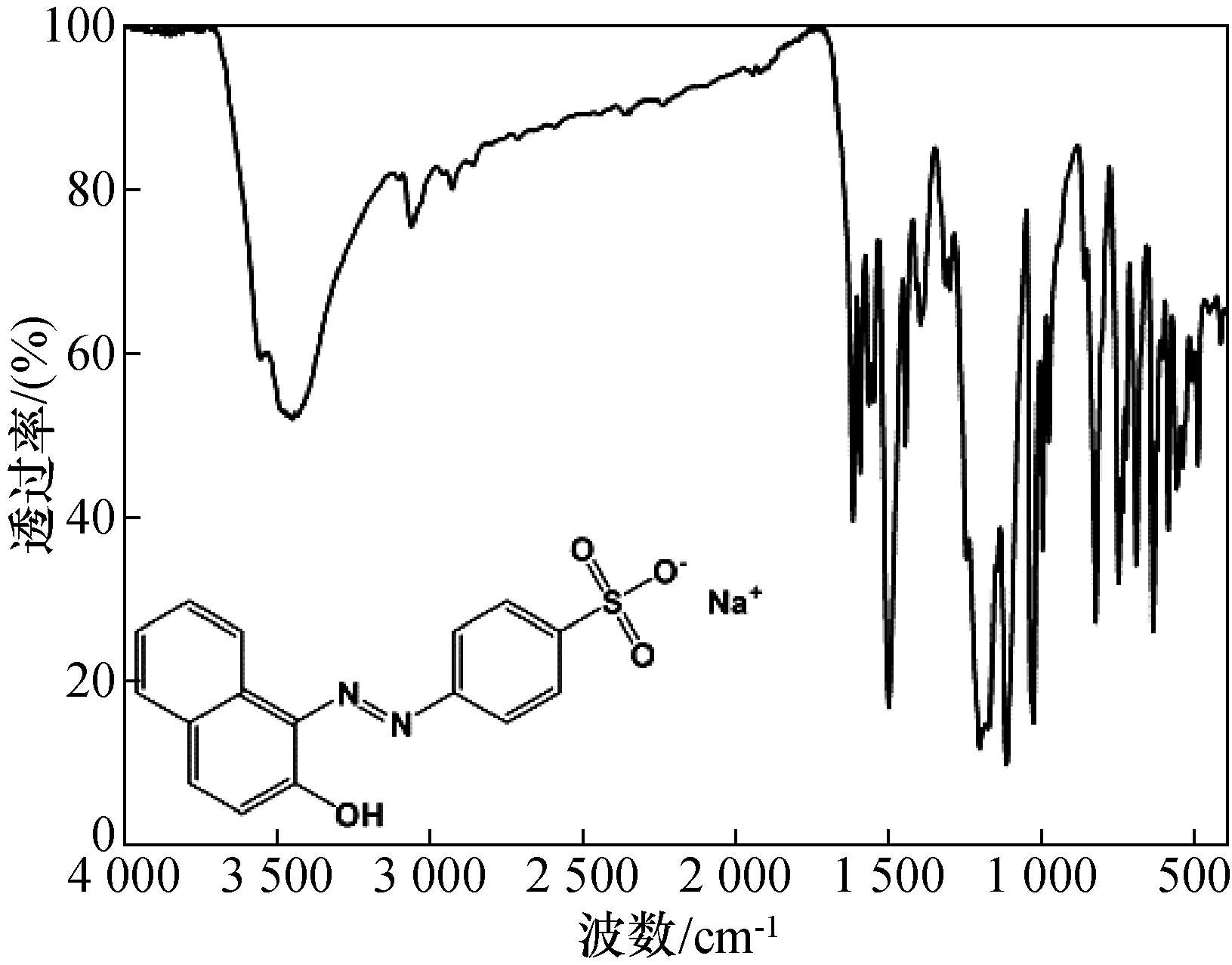
图1 酸性橙Ⅱ纯度候选标准物质的红外谱图Fig.1 Infrared spectrum of acid orange Ⅱ
图2给出了酸性橙Ⅱ纯度候选标准物质的核磁共振H谱,从化学位移上可以分析出H原子分别位于芳环C上及羟基O上。另外在高分辨质谱中可以发现质荷比m/z=327.044的特征峰(脱去Na+)。
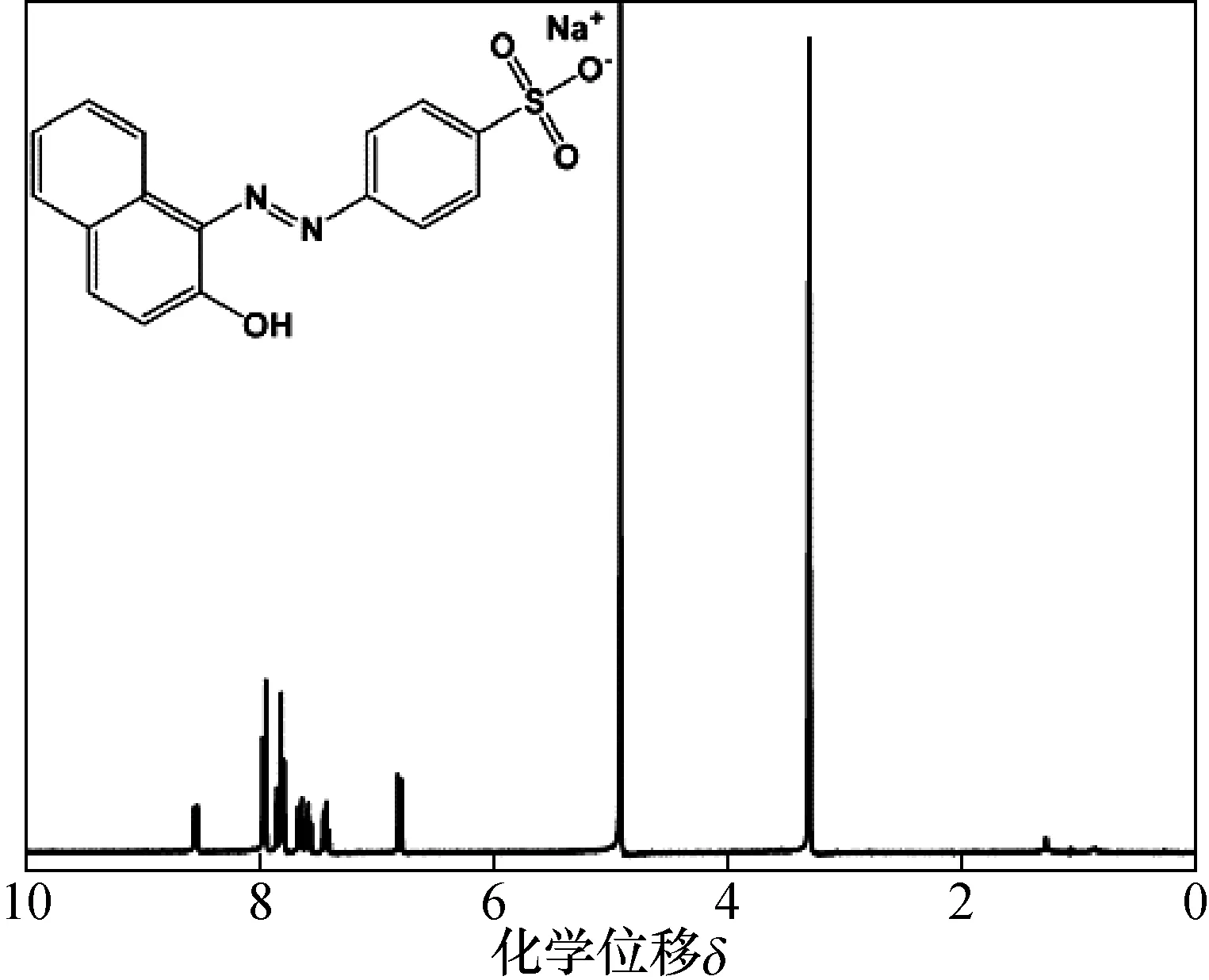
图2 酸性橙Ⅱ纯度候选标准物质的核磁共振H谱Fig.2 Nuclear magnetic resonance H spectrum of acid orange Ⅱ
3.3 均匀性检验
通常采用方差分析法来统计检验样品的均匀性。从已经分装好并编上号码的酸性橙Ⅱ纯度样品中按照头尾、中间编号各随机抽取15瓶样品。准确称量抽取的样品约10 mg,配制成溶液,采用液相色谱-面积归一化法,在95%的置信概率下采用方差分析考察样品的均匀性(ISO导则35),每瓶样品平行进样3次,3次测量的平均值作为均匀性评价的结果,然后对其进行方差分析,分析结果列于表1。

表1 酸性橙Ⅱ纯度标准物质均匀性结果方差分析Tab.1 The homogeneity and variance analysis results of acid orange reference material
表中:SS表示均值偏差的平方和;df为自由度;MS为平均方差;F为F值,是方差分析得到的统计量,用来检验回归方程是否显著;Fcrit为F临界值。
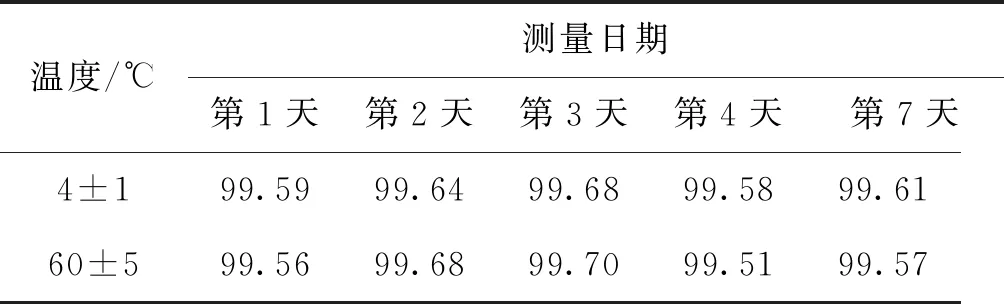
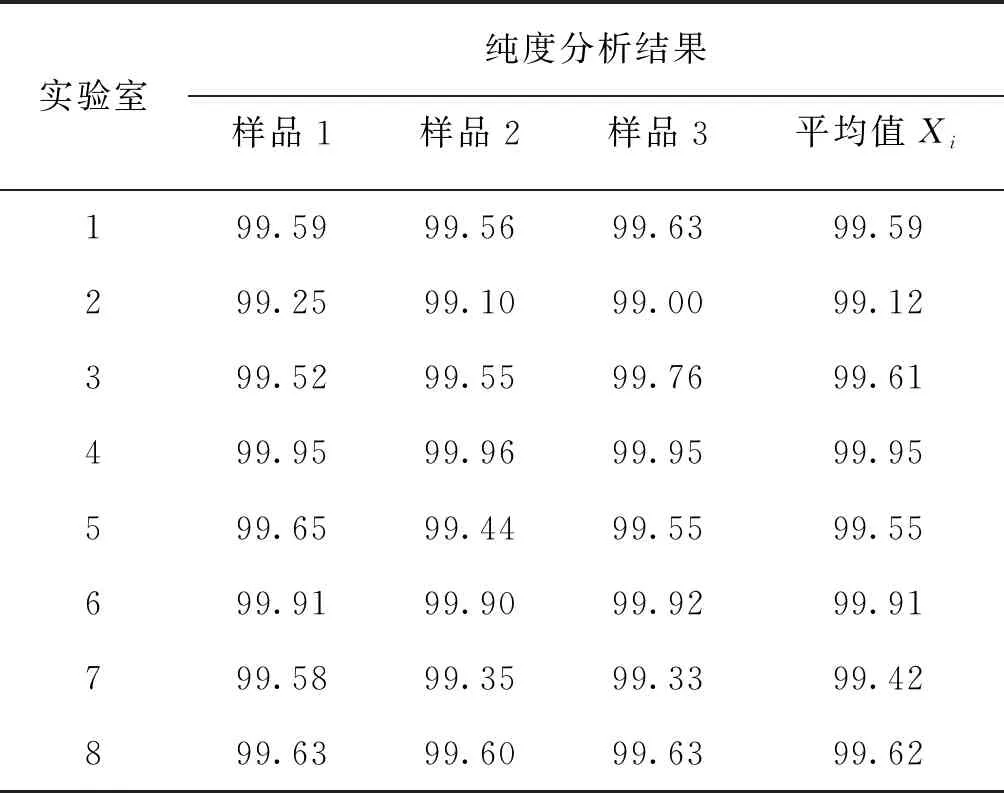
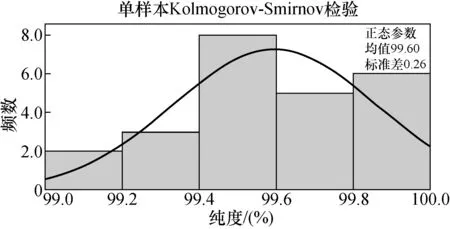
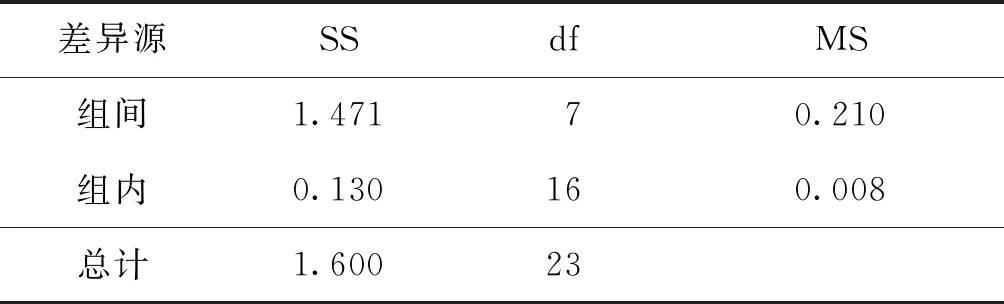
F 稳定性分为长期稳定性和短期稳定性。其中长期稳定性是指被定值的特性量随时间变化的情况,标准物质的长期稳定性受物理、化学和保存条件等因素的影响;而短期稳定性又称运输稳定性,是为了考察运输条件对标准物质的稳定性影响。 3.4.1 长期稳定性 按照前密后疏的原则,对酸性橙Ⅱ纯度标准物质稳定性进行了长期稳定性考察,为期30个月,实验数据见表2。 考察稳定性采用液相色谱法,应用面积归一化法,在不同的时间间隔内进行比对测量,每次随机抽取3瓶样品进行平行测量,取平均值作为稳定性考察结果,分析结果见表3。由表可知, |b1| 由于|b1| 表2 酸性橙Ⅱ纯度标准物质长期稳定性Tab.2 Long-term stability test of acid orange Ⅱ reference material 表3 酸性橙Ⅱ纯度标准物质长期稳定性数据方差分析结果Tab.3 Long-term Stability Variance Analysis Results of Acid Orange Reference Material 表中:b0为稳定性数据回归曲线的截距;b1为曲线的斜率;s(b1)为斜率的不确定度;t0.95,n-2为自由度是n-2和p=0.95(95%置信水平)时的t因子。 3.4.2 短期稳定性 为了考察运输条件对标准物质的稳定性影响,并综合考虑到南方的高温天气,实验模拟了运输条件,随机抽取6瓶样品,分别置于(60±5)℃的培养箱中(运输条件下)和(4±1)℃(常规储存条件下)保存一个星期,对比两种不同条件对其稳定性影响(以测量温度60 ℃为例)。 对两种条件下的测量数据进行同样的统计分析,分析结果见表4,可以发现两种条件下标准物质量值没有发生显著变化。 表4 酸性橙Ⅱ纯度标准物质短期稳定性数据方差分析结果Tab.4 Analysis of short-term stability variance of acid orange Ⅱ reference material (%) 通过长期稳定性和短期稳定性考察,在30个月的长时期内,酸性橙Ⅱ纯度标准物质稳定性良好,量值没有发生明显变化,该储存条件和运输条件很好地保证了标准物质的稳定性。 3.5.1 主成分纯度确定 酸性橙Ⅱ纯度标准物质采用质量平衡法定值纯度。联合8家实验室采用液相色谱紫外检测器法对主成分纯度进行测定。每家实验室仪器使用前均通过法定计量机构检定合格,各个实验室从全部样品中随机抽取3个包装,从每瓶中任意取出1~3个子样,用色谱级甲醇当溶剂配制测试液,使用液相色谱法测量酸性橙Ⅱ纯度标准物质。定值结果见表5。 表5 酸性橙Ⅱ纯度标准物质联合定值结果Tab.5 Characterization of acid orange purity reference material in 8 laboratories (%) 3.5.2 定值结果的检验 (1) 采用狄克逊准则来分析上述数据。按从小到大排列顺序为: 99.12<99.42<99.55<99.59<99.61<99.62<99.91<99.95 因n=8,则 (1) (2) 查表得D(a,n)=0.608,因此r8 (3) 查格拉布斯准则临界值G(0.05,8)=2.032, 1.825 (4) 因此,说明该组数据无异常值。 (3) 采用Kolmogorov-Smirnov检验方法在SPSS软件中对8家实验室定值数据进行正态分布检验,每家实验室3组数据,共有数据24组,结果如表6所示。 表6 8家实验室定值结果正态分布分析结果Tab.6 Normal distribution result of value data in 8 laboratories 图3为8家实验室定值数据正态分布图。从表6和图3可以看出:K-S检验中,渐近显著性(双侧)即P值为1.000,大于0.05,因此数据近似正态分布。 图3 8家实验室定值数据正态分布图Fig.3 Normal distribution map of value data in 8 laboratories (4) 采用科克伦(Cochran)法检查8家实验室数据之间是否为等精度。 计算8家实验室3次平行测量的标准偏差S,按从小到大排列,取最大值Smax,按公式(5)计算: (5) 计算得到C=0.358 5,此时m=8,n=3,α=0.05,查表得Cα=0.515 7;故C 故而可以采用8家实验室的总平均值作为联合定值的结果。总平均值如下: (6) 3.5.3 主成分含量核验 实验同时采用通用型电雾检测器(CAD)对酸性橙Ⅱ纯度进行验证。得到主成分含量>99.6%,与紫外检测器检测结果一致。 3.5.4 杂质含量测试 采用液相色谱-面积归一化法测定酸性橙Ⅱ的纯度时,结果并不包含标准物质可能含有的水分、溶剂残留和灰分,因此还需要进行杂质含量测试。 (1) 水分测定:酸性橙Ⅱ纯度物质水分测试利用卡尔费休水分测定仪,重复测量6次,采用狄克逊准则分析数据,发现无可疑数据。全部数据予以保留,取平均值0.19%作为水分测试结果(极差R=0.13%)。 (2) 残留溶剂测定:酸性橙Ⅱ纯品原料在加工过程中,易残留的溶剂为乙醇,采用顶空进样-气相色谱仪法对酸性橙Ⅱ和乙醇进行测试,发现在顶空条件下,酸性橙Ⅱ溶液中未检出乙醇等溶剂。 (3) 灰分测定:酸性橙Ⅱ纯度物质残渣通过高温灼烧测得,称取适量样品,在(650±50)℃的马弗炉中灼烧至恒重,称重测得结果,重复6次测量,采用狄克逊准则分析数据,发现无可疑数据。取平均值0.23%作为实验结果(极差R=0.15%)。 3.5.5 质量平衡法最终定值结果 根据质量平衡法即扣除杂质法: p=[100%-wnvi]×[100%-wrs-wwater-wash] =99.60%×(100%-0.19%-0%-0.23%) =99.2% (7) 式中:wnvi为非挥发性杂质的质量分数;wrs为残留溶剂的质量分数;wwater为水的质量分数;p为纯度;wash为灰分的质量分数;p为纯度。 根据《JJG 1006-94一级标准物质技术规范》的规定,标准值的总不确定度由3部分组成:第1部分是酸性橙Ⅱ定值实验引入的不确定度uw;第2部分是样品不均匀产生的不确定度ubb;第3部分是样品不稳定产生的不确定度uits[22~25]。 3.6.1 定值实验引入的不确定度 定值实验引入的不确定度由联合定值产生的不确定度及水分和残渣实验引入的不确定度等几部分组成。根据ISO Guide 35[3],多家实验室协作定值的不确定度可采用方差分析来进行估算。定值结果单因素方差分析得到的结果列于表7。 表7 定值结果单因素方差分析Tab.7 The variance analysis results of basic orange reference material 总平均值的不确定度如下计算: (8) 式中:sr为组内标准偏差;MSwithin为组内均方差。 (9) 式中:sL为组间标准偏差;MSamong为组间均方差;n为测量次数。 (10) 式中:uLC为液相色谱法定值引入的不确定度。 而根据水分测量的数据,测定的R=0.13%,n=6(查表c=2.53),采用极差法计算水分引入的不确定度分量为:uwater=R/c=0.05%。根据灰分测量的数据,测定的R=0.15%,n=6(查表c=2.53),采用极差法计算灰分引入的不确定度分量为:uash=R/c=0.06%。最终: (11) 3.6.2 均匀性引入的不确定度 均匀性采用酸性橙Ⅱ的纯度为比较参数,样品瓶间不均匀性所产生的标准偏差必须并入到总的不确定度中。根据表1均匀性分析结果可知,样品间不均匀的标准偏差为: (12) ubb≈0.067 3% 3.6.3 稳定性引入的不确定度 根据酸性橙Ⅱ长期稳定性的实验数据及回归方差结果可知分由稳定性结果分析可知,有效期t=30 月的长期稳定性的不确定度贡献即为: uits=s(b1)·t=8.1×10-3×30=0.243% (13) 3.6.4 合成扩展不确定度 合成各部分不确定分量: =0.282% (14) 扩展不确定度: U=k×u=2×0.282%=0.56%≈1.0% (15) 酸性橙Ⅱ纯度标准物质的定值结果为:W=(99.2±1.0)%,k=2。 研制标准物质过程中,需对定值结果或测量方法进行国际、国内比对或测量验证工作。目前,国内外酸性橙Ⅱ标准品主要有纯度和溶液两种类型 ,多用于食品、化妆品等产品中酸性橙Ⅱ的检测。表8列出了部分国内外酸性橙Ⅱ标准物质情况及比对结果。 表8 酸性橙Ⅱ纯度和溶液同类标准物质的情况对照表Tab.8 Comparison of purity and solution of acid orange Ⅱ reference materials 选用Dr. Ehrenstorfer®GmbH 的C1573500与本文GBW(E)100373标准物质进行比对分析,将两种纯度物质配制相应浓度的溶液进行液相色谱纯度分析。结果表明使用进口的纯度标准品单点校正测定广东省计量科学研究院研制的酸性橙Ⅱ纯度标准物质,其值均落在定值的不确定度范围,因此GBW(E)100373酸性橙Ⅱ纯度标准物质量值是可信的,且具有稳定、便捷的特点。 以经典的质量平衡法研制了GBW(E)100373酸性橙Ⅱ纯度标准物质,重点介绍了研制过程中标准物质样品的制备、均匀性检验、稳定性检验、定值、不确定度评定及比对和验证,是食品成分中有机成分分析标准物质研制的一个典型代表。 酸性橙Ⅱ纯度标准物质可以满足食品、医药、日常化学品、环保、化学产品研究和测试需求,也将用于对相关仪器(例如比色板、荧光分光光度计等)校准和分析方法的评估。未来,期待研制出更多食品成分分析标准物质,促进食品及食品添加剂行业的健康有序发展,为保障广大消费者的健康而做出贡献。3.4 稳定性检验

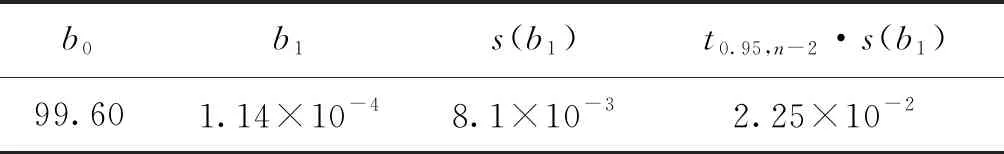
3.5 定值

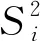
3.6 不确定度的评定
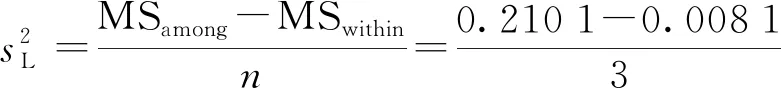
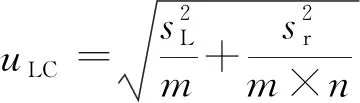

3.7 标准物质的比对和验证

4 结 论
