基于16S rRNA高通量测序的鳢肠道微生物群落结构研究
2020-10-13徐晟云陈昆慈罗青刘海洋欧密上官清赵建
徐晟云,陈昆慈,罗青,刘海洋,欧密,上官清,赵建*
(1.中国水产科学研究院珠江水产研究所 农业农村部热带亚热带水产资源利用与养殖重点实验室,广东 广州 510380; 2.上海海洋大学 水产与生命学院,上海 201306)
鳢是中国重要的优质经济鱼类之一,2014年全国产量达51万t[1],养殖品种以乌鳢Channaargus、斑鳢Channamaculata和杂交鳢为主。其中,乌鳢生长快、个体大、抗寒能力强,主要在中国北方地区养殖;杂交鳢是以乌鳢和斑鳢为亲本杂交获得的养殖品种,表现出显著的杂交优势,并能摄食人工配合饲料,是长江流域及以南水域主要养殖品种。近年来,中国水产科学研究院珠江水产研究所培育出的新品种乌斑杂交鳢C.argus♀×C.maculata♂,以乌鳢为母本、斑鳢为父本,其具有较强的抗寒能力,在黄河流域及以北地区得到广泛推广[2]。
鳢具鳃上器,能够呼吸空气,适合高密度养殖,近年来,随着养殖密度的不断提高,其养殖过程中病害发生率高居不下,因此,提高鳢自身免疫能力及抗病能力已成为养殖过程中备受关注的一个课题。肠道微生物被誉为宿主的一个附加“器官”[3-4]。在肠道群落结构中,微生物的失衡可能会阻碍宿主生理活动的进行,继而引发疾病等。在鱼类肠道里,有一些微生物会在形成过程中逐渐演变成固有群落,而有一些则形成了非固有菌群[5]。肠道微生物群落结构特征组成和多样性会影响营养物质加工、消化,维持宿主能量平衡、免疫疾病和生长发育等生理活动[6-7],研究者通过选择不同饵料品种及适当的饵料添加剂,促使养殖对象的肠道菌落结构特征发生变化并朝着有益的方向发展[8],从而提高养殖动物的生长和健康水平。因此,研究鱼类肠道微生物群落结构对于鳢的健康生态养殖有着极其重要的作用[9]。
本研究中,采用Illumina高通量测序技术对鳢16S rRNA基因进行测序并分析其肠道微生物组成,旨在探讨不同环境下鳢肠道内微生物群落结构特征,为研究鳢生长、疾病与肠道微生物结构特征间的关系,以及促进鳢的健康养殖提供科学依据。
1 材料与方法
1.1 材料
2018年10—11月分别在珠海斗门(DM)、佛山南海(百容水产良种有限公司的水泥池BRSNC和池塘BRCT)、佛山顺德杏坛(XT)、阳江(YJ)4个地区共5个点采集成年健康有活力的鳢样品,无菌环境下解剖取其肠道内容物后保存于液氮中带回实验室,共31个样品,其中,阳江的样品为野生斑鳢(50~100 g),其他样品均为杂交鳢(100~150 g)。样品信息见表1。
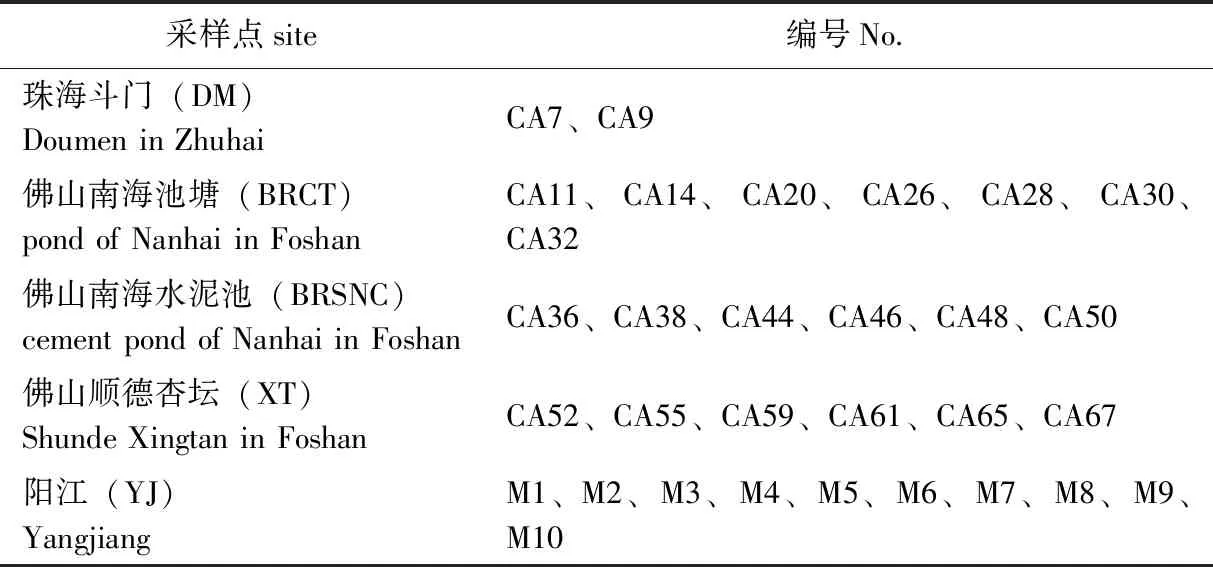
表1 样品信息Tab.1 Sample information
1.2 方法
1.2.1 样品基因组提取 采用CTAB法提取总样品的DNA[10]:在装有0.3 g样品的离心管中加入1 mL 65 ℃条件下预热的CTAB提取液和20 L蛋白酶K,65 ℃条件下烘箱温浴30 min,之后以8000 r/min离心5 min。取上清液800L至2.0 mL无菌离心管中,加入800 L氯仿/异戊醇(24∶1),以12 000 r/min离心20 min。吸取上清液600 L至2.0 mL无菌离心管中,加入等体积氯仿/异戊醇(24∶1),以12 000 r/min离心20 min。再吸取上清液400 L至1.5 mL无菌离心管中,加入2/3体积的异丙醇和1/10体积的乙酸钠,充分混匀,于-20 ℃下放置1 h。以12 000 r/min离心10 min,弃上清,瞬时离心,离心管开盖,在37 ℃条件下烘箱晾干10 min至DNA沉淀呈半透明状。加入100 L无菌ddH2O,溶解DNA沉淀,37 ℃下水浴锅消化1 h后,-20 ℃条件下保存,送至北京迈克生物科技有限公司完成16S rRNA基因扩增子测序。
1.2.2 16S rRNA 基因扩增子测序 对DNA样品中16S rRNA基因v3~v4区域进行测序及信息分析。采用通用引物338F:5′ACTCCTACGGGAGGCAGCA 3′和806R:5′GGACTACHVGGGTWTCTAAT 3′用于扩增16S rRNA基因的v3~v4区域进行Illumina深度测序。PCR反应体系(20L):ddH2O 13.25 μL,DNA模板(100 ng/mL) 0.5 μL,10×PCR ExTaq Buffer 2.0 μL,Prime 1 (10 mmol/L) 1.0 μL,Prime 2 (10 mmol/L) 1.0 μL,dNTP 2.0 μL,ExTaq (5 U/mL) 0.25 μL。反应条件为:95 ℃下预变性5 min;95 ℃下循环变性30 s,58 ℃下退火复性20 s,72 ℃下延伸6 s,共进行30个循环;最后在72 ℃下再延伸7 min。然后取5 μL PCR产物使用10 g/L琼脂糖凝胶电泳法纯化和回收扩增产物。使用Qubit 2.0荧光计测定浓度并连接接头完成文库构建。检测文库片段范围和浓度后,选择Illumina HiSeq 2500平台对文库进行测序。
1.2.3 信息分析流程 根据PE reads间的重叠关系,将测序得到的双端序列数据组成一条序列Tags,同时过滤reads的质量和合并效果。使用FLASH 1.2.7软件拼接重叠各样品的reads,获得原始的数据(Raw Tags);使用Trimmomatic 0.33软件过滤不合格的原始数据,得到合格的数据(Clean Tags)后,使用UCHIME 4.2软件鉴定识别嵌合序列同时去除不合格的杂质,得到有效数据(Effective Tags)。
1.2.4 生物信息学分析
(1)OTU分析。OTU(Operational Taxonomic Units)是人为给某一个单元(品系、种、属等)设置的分类标志。在相似性 97% 的水平上,使用QIIME 1.8.0软件中的 UCLUST聚类Tags,划分操作分类单元[11]。
(2)Alpha 指数分析。 Alpha 多样性是指一个特定区域或生态系统内的多样性,是反映丰富度和均匀度的综合指标。群落丰富度的指数主要包括Chao1指数和ACE指数;群落多样性指数主要包括Shannon指数和Simpson指数。其中,Chao1指数越大,表明群落的丰富度越高;ACE指数越大,表明群落的丰富度越高;Shannon指数值越高,表明群落的多样性越高;Simpson 指数值越大,说明群落多样性越低[12]。Alpha指数可用Mothur 1.30软件分析得出。
(3)稀释性曲线分析。稀释性曲线[13]是从样本中随机抽取一定数量的个体,统计这些个体所代表的物种数目,并以个体数与物种数来构建曲线。当曲线趋向平坦时,说明测序数据量合理。利用Mothur 1.30软件进行Rarefaction 分析,利用Perl 语言工具制作曲线图。
(4)热图分析。 Heatmap[14]通过颜色梯度及相似程度来反映多个样品在各分类水平上群落组成的相似性和差异性。利用R语言的Vegan 包、Vegdist和Hclust 进行距离计算和聚类分析;用Chao算法计算距离,Complete方法聚类,将聚类后数据表示在Heatmap 图上。
(5)PCA分析。 主成分分析 (Principal Component Analysis,PCA)[15]运用方差分解,将多组数据的差异反映在二维坐标图上,两个样品距离越近,则表示这两个样品的组成越相似。使用R语言工具分别绘制PCA分析图。
2 结果与分析
本试验中31个样品测序共获得2 636 884对reads,拼接、过滤后共产生1 768 461条合格序列(Clean Tags),每个样品至少产生25 112条合格序列。
2.1 鳢肠道微生物物种多样性
从表2可见,4个地区5个采样点共31个样品之间具有的OTU数量不同,其中CA50样品OTU数最多(359个),M8样品OTU数最少(34个),本试验样品中获得微生物总OTU数为585个。
将4个地区样品间共有及特有OTU数目通过Venn图展示。从图1可见:4个地区5种不同环境共同拥有的OTU数为119个,占4个地区总OTU数的20.34%,分别占佛山南海池塘OTU总数的23.66%(OTU总数503)、佛山南海水泥池OTU总数的28.23%(OTU总数421)、杏坛OTU总数的30.13%(OTU总数395)、阳江OTU总数的42.5%(OTU总数280)、珠海斗门OTU总数的48.77%(OTU总数244)。
2.2 鳢肠道微生物群落结构多样性差异
2.2.1 稀释曲线结果 从图2可见:抽样reads在10 000以下时,随着测序条数的加大,每一条曲线起始时表现为急剧上升,这表明鳢肠道内容物中有大量不同菌群被发现;在10 000~30 000时,曲线的序列数越来越趋于平缓,表示该环境的物种未检测出来的测序数量不会再显著增加,即此次检测的微生物已近乎饱和,更多的测序量对发现新的OTU边际贡献较小,本测序数据量合理,反映出样品多样性的完整度;但当抽样reads超过30 000时曲线略微上升,可能是因为某些原因致使肠道中残留了少许的菌群或少数细菌死亡后未被检测出[16]。
2.2.2 肠道微生物Alpha多样性 从表2可见:每个样品的Coverage值均达到了99%以上,这说明每个样品序列几乎全被检测出且达到饱和状态,OTU覆盖率数值越高,样本物种检测概率则越高。Coverage值可反映此次样品中肠道微生物菌群结构组成及多样性的真实性,每个样品的Coverage值均接近百分百,证实了此次试验数据的可靠性。

表2 OTU数及Alpha多样性指数统计Tab.2 Statistics of OTU number and Alpha diversity index
比较31个样品的Alpha多样性发现:CA50的Chao1值最大,高达364.181 8,说明这个样品里的微生物菌群丰富度最大;CA44的Simpson值最小,为0.018 5,Shannon值最大,为4.522 2,说明此样品的肠道微生物群落多样性最大。
2.3 鳢肠道微生物群落组成分析
从表3可见:佛山南海池塘养殖的鳢肠道内容物中,优势菌群主要为红球菌属Rhodococcus(12%)、鲸杆菌属Cetobacterium(7%)、梭菌属(1.5%)、微杆菌属Aurantimicrobium(5%)、邻单胞菌属Plesiomonas(0.5%)、未培养的细菌Mycoplasmataceae(20.5%);佛山南海水泥池养殖鳢的肠道内容物中,优势菌群主要为弧菌属Vibrio(30%)、微杆菌属(5.5%)、红球菌属(5.17%)、梭菌属(1.5%)、鲸杆菌属(0.2%)、未培养的细菌Mycoplasmataceae(2%);珠海斗门池塘养殖鳢肠道中,优势菌群主要为梭菌属(79%)、红球菌属(4%)、微杆菌属(3%)、气单胞菌属Aeromonas(2%);佛山顺德杏坛池塘养殖鳢肠道中,优势菌群主要为鲸杆菌属(27%)、梭菌属(22.2%)、红球菌属(3%)、弧菌属(0.5%);阳江野生斑鳢肠道中,优势菌群主要为鲸杆菌属(40.5%)、梭菌属(22.5%)、邻单胞菌属(20%)、气单胞菌属(8%)、红球菌属(3.1%)。
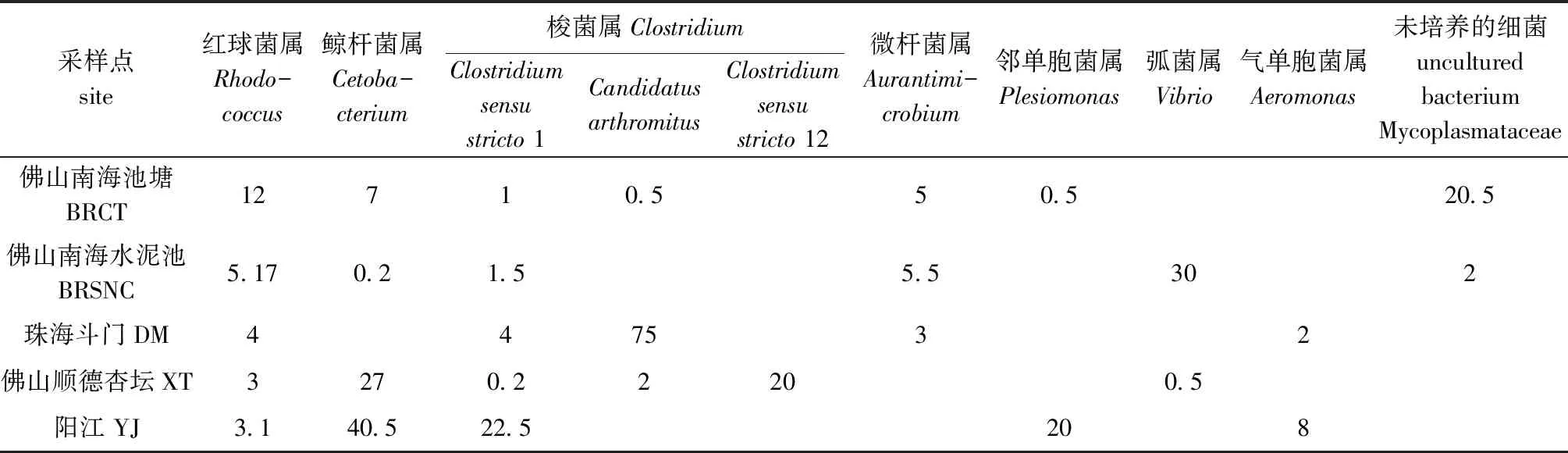
表3 属水平上鳢肠道微生物优势菌群占比Tab.3 Microbial dominant flora in intestine of snakehead at the genus level %
根据31个样品的微生物组成和相对丰度进行物种热图分析,更加详细地表述了不同地区的优势菌群具有明显差异。从图3可见:佛山南海水泥池(BRCT)养殖鳢肠道的优势菌群主要有肠球菌属Enterococcus、乳杆菌属Lactobacillus、拟杆菌属Bacteroides、Prevotella(属于拟杆菌属)、Bosea(属于变形杆菌属Proteus)、Phreatobacter(属于变形杆菌属)、Rhodobacter(属于变形杆菌属)等;而佛山南海池塘养殖鳢肠道中的优势菌群则为棒杆菌属Corynebacterium、短杆菌属Brevibacterium、Cloacibacterium(属于拟杆菌属)、梭菌属、Massilia(属于变形杆菌属)、罗斯氏菌属Rothia等;珠海斗门(DM)养殖鳢肠道微生物优势菌群相对集中,只有弧菌属和梭菌属;佛山顺德杏坛(XT)养殖鳢肠道的优势菌群有假单胞菌属Pseudomonas、泛菌属Pantoea、假单胞菌属Acinetobacter、短波单胞菌属Brevundimonas等;阳江(YJ)采集的野生鳢肠道内容物中,优势菌群有葡萄球菌Staphylococcus、梭菌属、气单胞菌属等。热图分析发现,鳢肠道微生物群落中还包含了变形杆菌属、肠球菌属、乳杆菌属、拟杆菌属、棒杆菌属等其他菌属,但是占比不高,说明菌群数量不多。
2.4 微生物群落结构差异分析
对菌群结构进行主成分分析,结果显示:第三象限中有佛山南海水泥池(BRCT)、池塘(BRCT),以及佛山顺德杏坛(XT)养殖鳢、阳江的野生鳢、肠道微生物群落形成一个组群,说明这些地区鳢肠道微生物群落结构差异较小;第二象限中只有珠海斗门(DM)样品;第四象限中包含佛山南海池塘的个别样品及佛山顺德杏坛的个别样品;第一象限中全部为阳江(YJ)的野生斑鳢(图4)。PCA分析发现,人工养殖杂交鳢的肠道微生物群落结构比较接近,与野生鳢的群落结构具有一定差异。
3 讨论
3.1 不同环境下的微生物多样性和丰度
本研究中不同环境中鳢肠道内容物样品间OTU个数具有一定的差别,总数达到585个,共同拥有的OTU数为119个。李存玉[17]分析池塘养殖牙鲆肠道菌群结构及其与益生菌调控的关系时发现,两种养殖模式下牙鲆肠道样品OTU总数为367个,共有OTU数为270个。黄丽丽等[18]比较冷水鱼样品时发现,总共聚成242个OTU。秦亚玲等[19]比较3处热泉(碱性、酸性、中性)发现,分别聚类形成141、79、58个OTU。与上述研究相比,本研究中OTU总数更大,表明鳢肠道中微生物物种丰度更大,这可能是鱼类肠道微生物数量和群落结构与其生存的水环境及摄食条件不同等因素相关[20-21]。厚壁菌门为大多数脊椎动物肠道中的优势菌群[22],而厚壁菌门、变形菌门、放线菌门、拟杆菌门等为鱼类消化道中常见的微生物优势菌[23]。
3.2 不同环境下的优势菌群及其作用
通常情况下,淡水鱼消化道中常见的微生物有气单胞菌属、邻单胞菌属、肠杆菌属、假单胞菌属等,而海水鱼消化道中常见的则为弧菌属、假单胞菌属、不动杆菌属等[24]。Huber等[25]使用了分子生态学方法对虹鳟Oncorhynchusmykiss肠道微生物群落进行研究,获得了一些常规培养方法下不能发现的优势细菌,如肉杆菌Carnobacteriumcollins、肉毒杆菌Clostridiumbotulinum、硫酸盐还原菌Sulfate-Reducingbacteria等。杨桂梅等[26]对用不同饲料投喂的暗纹东方鲀Takifuguobscurus的表皮、性腺和肠道进行研究分析,发现这些样品中的菌落组成以弧菌为主。李学梅等[27]研究了3种常见养殖鱼类的肠道微生物,其中斑点叉尾鮰肠道中的优势菌群为变形杆菌,而异育银鲫和银鲫肠道中优势菌群分别为梭杆菌属和气单胞菌属。本研究样品中优势菌群主要为红球菌属、鲸杆菌属、梭菌属、微杆菌属、弧菌属、气单胞菌属、邻单胞菌属等,其中,红球菌属属于放线菌门,梭菌属属于厚壁菌门,这与国内外学者研究结果相似,不同的是本研究中优势菌群并未出现肠杆菌属、肉杆菌属等微生物,这说明肠道微生物菌群结构会受到外界环境的影响而有所不同。
不同环境下的优势菌群存在显著性差异,梭菌属、红球菌属虽在所有样品中均存在,但不同环境下所占比例不同,说明肠道微生物的组成受到环境的影响,这与Ni等[28]利用DGGE技术比对不同环境草鱼的肠道微生物菌群结构,发现不同生活环境的草鱼肠道微生物群落结构不同的结果相似,李存玉等[29]通过比较分析池塘和工厂化养殖牙鲆肠道菌群结构时也发现,两种养殖模式下牙鲆肠道中的优势菌种类相同,但同种优势菌的数量分布存在较大差异。本研究中优势菌群不同也可能与宿主摄食饲料不同等因素息息相关,已有研究表明,食性是影响鱼类肠道微生物群落结构的一个显著因素[30]。Ward等[31]发现,食性对鱼类肠道微生物菌群结构特征有显著影响,Ingerslev等[32]和Ni等[33]的研究结果也表明,食性是影响肠道微生物菌落的主要因素之一。
此外,梭菌属细菌可利用发酵碳水化合物和糖类为鱼类提供机体生存所需能量,以促进生长、增强免疫功能和抗癌功能[34-35]。梭菌属中的丁酸梭菌能促进拟杆菌的繁殖,拟杆菌属细菌能利用多糖和释放抗炎物质以增强机体免疫系统的免疫能力,目前梭菌属细菌已作为益生菌广泛运用在鱼类养殖中[36]。鲸杆菌属可发酵多肽碳水化合物以及合成维生素B12[37],红球菌能够利用有机化合物作为碳源和能源[38],微杆菌细菌能抑制病原菌的活性,可用来生产抗菌剂[39]。
本研究中红球菌属、梭菌属、微杆菌属、拟杆菌属等存在于鳢肠道中,其作为优势菌群可以促进鳢对营养物质的消化吸收,并抵御病原菌的入侵,对机体的生长发育和健康起到重要作用,属于有益菌。本试验通过对不同环境下鳢肠道微生物种群结构分析,获得了正常状态下鳢肠道微生物组成,为鳢肠道菌群调节、开发相关产品,促进鳢健康养殖提供了数据参考。
4 结论
(1)不同环境的鳢肠道微生物类群数量和种类存在显著差异。
(2)相同环境的鳢肠道微生物群落结构较为接近,即人工养殖鳢的肠道微生物群落结构较为接近,而与野生鳢之间肠道微生物群落结构差异明显。
