基于合成生物学策略的酶蛋白元件规模化挖掘
2020-10-10张建志付立豪唐婷张嵩亚朱静李拓王子宁司同
张建志,付立豪,唐婷,张嵩亚,朱静,李拓,王子宁,司同
(中国科学院深圳先进技术研究院,深圳合成生物学创新研究院,中国科学院定量工程生物学重点实验室,广东深圳518055)
酶(enzyme)是一种重要的生物催化剂,可以促进生物体内化学反应在生理条件下高效和特异地进行,其化学本质是蛋白质或RNA[1]。酶蛋白具有催化效率高、专一性强、作用条件温和、环境友好等特点,被用于开发工业酶制剂和构建微生物合成代谢途径[2-4],在医药、化工、能源、材料等领域有广泛应用[5-7]。例如,研究者从罂粟、褐鼠、假单胞菌、黄连和花菱草中挖掘酶蛋白,构建了包含18个酶催化步骤的合成途径,实现了抗癌化合物诺司卡品在酿酒酵母中的高效生产[8-9]。酶蛋白具有天然多样性,截至2019年,NCBI(National Center for Biotechnology Information)数据库中共有约4×108种特异性的蛋白序列;且随着DNA测序技术的快速发展使得蛋白序列数量每24个月增加约1倍[10-12]。在此背景下,对酶蛋白资源进行规模化的挖掘和开发变得极为重要和迫切。
传统酶资源挖掘一般采用菌种筛选、功能宏基因组等技术。菌种筛选以催化活性为导向,首先对天然微生物进行培养与筛选,再通过分子生物学技术克隆与鉴定相关基因,或者对微生物蛋白组进行提取、分离与组学分析,鉴定活性酶蛋白;功能宏基因组方法基于活性或DNA序列相似性,对表达宏基因组DNA片段的克隆进行筛选,用于挖掘环境中的未培养微生物基因资源。两类方法都依赖实体样品,研发周期长,成本高,具有盲目性。
相较于传统方法,合成生物学为酶蛋白的规模化研究提供了新的思路。基于工程学理念,合成生物学对生物体进行有目标的“设计—构建—测试—学习”,获得具有特定功能的人工生命系统。就酶催化系统而言,合成生物学采用“自下而上”的策略,首先针对目标生化反应,从数据库中识别酶蛋白的氨基酸序列;接着根据底盘细胞适配原则,设计并合成酶元件、调控元件的DNA序列,利用标准化DNA组装方法构建蛋白表达模块或合成代谢通路;最后在底盘细胞中进行转化、表达与功能表征。与传统方法相比,基于合成生物学进行酶蛋白挖掘具有诸多优点,如不受生物实体的限制,可以利用生信方法针对性确定研究对象,提高新颖元件的开发效率;DNA合成可以进行序列工程化改造,从密码子偏好、亚细胞定位等方面提高酶元件与底盘的适配性;基于标准化底盘、流程和实验条件开展研究,有利于积累优质元件与数据,为人工智能指导的酶元件挖掘等研究方法奠定基础。
利用合成生物学对酶蛋白进行工程化挖掘,需要开发高通量的计算和实验方法。目前缺乏从序列准确预测功能的方法,往往需要筛选大量候选序列,才能够识别自然界中催化目标生化反应的酶蛋白;同时,天然酶元件往往需要进行工程改造才能达到特定指标,如催化效率、专一性、底盘细胞适配性等。因此,需要基于合成生物学理念开发工程化、标准化的技术和流程,从海量测序数据中识别、合成、表征天然酶元件。针对酶元件的识别,传统方法基于同源序列比对、蛋白质家族结构域比对等原理进行酶功能注释。但是,序列相似性与功能之间并不总是完全对应,例如序列同源性达到98%的蛋白质可能具有不同的生化功能[13],而序列同源性很低的蛋白质也可能具有相同的生化功能[14-15]。因此,需要开发新的生物信息方法,对酶蛋白从序列、三维结构、进化关系和蛋白互作等多个层面进行综合性分析,根据实验目的优先化排序用于功能验证的候选序列。另外,不同来源酶蛋白在底盘细胞中进行异源表达时,可能存在密码子偏好不同、无法折叠、稳定性低、辅酶因子及底物缺失等适配问题,需要进行工程化实验设计(design of experiment,DOE),对DNA序列、调控元件、DNA组装方法、蛋白表达条件等因素进行系统性探索和优化。综上所述,对上百个候选序列的功能表征和对实验条件的系统性优化,对研究平台的操作通量和自动化水平提出了新的需求。
本文作者对利用合成生物学方法高通量挖掘酶元件的研究进展进行综述。介绍了相关的生信工具,已有文献对多序列比对(multiple sequence alignment,MSA)、基因注释、同源建模(homologous modeling)等内容进行了系统总结[16-17],本文着重关注聚类分析、可溶性预测、杂泛性预测等算法。总结了对候选酶元件进行合成组装、异源表达、功能筛选的高通量实验技术。接着,讨论了规模化挖掘技术在酶家族系统性研究方面的应用。搭建自动化合成生物学平台开展酶蛋白高通量研究是未来的趋势。
1 计算机辅助设计用于高通量酶元件挖掘
高通量挖掘酶元件的一般流程如图1所示[18]。随着测序技术的飞速发展,大量(宏)基因组和转录组得到解析,从中可以预测得到大量蛋白序列。与蛋白质结构域数据库进行同源比对可以实现酶功能的初步注释[19]。同时,代谢多样性提供了丰富的生物催化资源,各类初级和次生代谢产物合成途径的预测和分析,也为酶元件的挖掘提供了帮助。但是,目前很难从酶序列出发准确预测其催化活性、底物选择性、可溶表达等关键性质,需要开发新的算法对候选元件进行优先化排序,提高功能筛选的成功率,利用最少的实验资源探索相似酶蛋白的功能多样性。下面将从酶的聚类分析(clustering)和实验验证的优先化排序两个方面进行介绍。
1.1 酶蛋白聚类分析
各种类型的蛋白质资源数据库为酶元件的高通量挖掘提供了宝贵的材料,如表1所示。截至2020年2月,蛋白质资源数据库UniProt中储存了约18亿条蛋白质序列。其中大部分功能注释是通过与CDD(conserved domain database)[20],Pfam[21],CATH[22]和FIGfams[23]等数据库中蛋白的结构域序列进行同源性比对而获得[19];但至少50%的注释并不精确甚至是错误的[24]。因此,需要整合不同层次的功能注释工具和实验数据,帮助科研人员从序列、结构、进化和蛋白互作等多个层面对候选酶元件进行综合分析。其中,酶蛋白聚类分析可以利用可视化手段最大程度地利用文献中实验数据对未知蛋白质进行多个维度的功能注释[25]。下面将重点介绍用于酶蛋白聚类分析的两个方法:序列相似性网络分析工具(sequence similarity network,SSN)[26],以及侧重蛋白质结构比对的CATH分析工具(Class,Architecture,Topology,Homologous superfamily)[22]。

图1 酶蛋白资源高通量挖掘流程图Fig.1 Workflow for high-throughput enzyme mining
SSN是一种可显示同源蛋白之间两两序列相似性关系的多维网络[27]。网络中每个蛋白质由一个节点(node)表示;如果两个蛋白质间具有超过序列相似性(blastE-value)的指定阈值,则代表它们的两个节点通过一条边(edge)连接;每个簇(cluster)中的节点至少有一条边与簇中的其他节点相连。选择合适的阈值是成功构建SSN的关键。SSN中的“节点属性”包含每个节点的各种信息,比如系统分类,TrEMBL/SwissProt、PDB和GO等数据库信息的链接[19,28-29]。这些信息有助于用户对SSN进行分析,通过设定合适的阈值将不同节点聚类。与多序列比对和系统进化树(phylogenetic tree)方法相比,SSN能够快速地分析更大的蛋白质序列集,并能同时评估正交信息。例如,映射到序列相似性的功能多样性,有助于对酶家族中共享显著序列相似性的子群如何关联进行推测。SSN分析也可揭示序列相似性较低但具有相似结构或功能的远同源蛋白之间的关系,从而指导研究者优先探索序列空间内的未知区域[27,30]。最初,Barber等[31]利用Python语言编写了Pythoscape工具用于生成SSN;但该工具需要基于服务器集群在Unix环境下执行命令行脚本,并未得到广泛的应用。为了推广SSN的应用,研究者开发了用户友好的EFI-EST网络工具(enzyme similarity tool)[25,32],可以通过浏览器访问(https://efi.igb.illinois.edu/efi-est/),使得用户不需要专业编程知识即可快速生成SSN,其结果可以在开源的Cytoscape软件中进行可视化分析。
CATH工具可以揭示蛋白质结构域间的进化关系。基于空间结构的相似性,CATH识别wwPDB数据库(Worldwide Protein Data Bank)中的蛋白质结构域,并将其聚类形成结构域超家族(domain superfamily)[22]。由于仅有少数蛋白质(10万余种)具有实验测定的三维空间结构信息,CATH-Gene3D工具将具有代表性的结构域转换成隐性Markov模型,形成基于一级序列的“指纹图谱”库;利用这一方法,共预测得到43万个蛋白结构域,构成6000余个结构域超家族。CATH进一步按照Class(二级结构的组成)、Architecture(二级结构形成的形状)和Topology(二级结构连接的顺序)的三个层次,分析结构域超家族间的进化关系[33]。另外,FunTree数据库进一步将酶蛋白的功能信息,如生化反应物的结构、酶催化动力学数据等,与CATH结构域超家族分类进行整合,从而综合分析序列、结构、功能和进化关系。该数据库目前已包含2340个结构域超家族、7万个结构域和40万个代表性序列[34]。

表1 酶元件资源数据库及链接Tab.1 Enzyme resource database and their corresponding links

续表

续表
1.2 实验验证的优先化
聚类分析对海量蛋白质数据库进行初步筛选之后[35],仍存在大量候选序列,远远超出实验验证的能力[18]。例如,软海绵素B合成途径包含47步不同的酶催化反应,假设每个步骤候选酶数量为100个,则需要构建和筛选的代谢途径组合可达到约1094个[36]。因此,需要探索优先化标准与算法对候选酶进行排序,提高功能验证实验的效率[2,4]。
酶蛋白的可溶性表达和底物杂泛性(promiscuity)是优先化排序的重要标准。可溶性表达是对酶进行功能表征的前提,目前已有一些基于能量计算、机器学习和进化分析的算法对这一性质进行预测[37]。例如,Wilkinson-Harrison溶解度模型可以预测蛋白序列在大肠杆菌中可溶性表达的概率[38]。Vanacek等[18]对脱卤酶家族进行生信分析及优先化选择之后,筛选得到20个候选蛋白序列进行表达实验,最终有60%的蛋白质在大肠杆菌中实现可溶性表达,与模型预测得到的理论值一致。另外,需要根据实验目的选择具有不同底物杂泛性的酶。体外生物催化体系通常使用单一底物,使用高底物杂泛性酶不影响反应专一性,并且有利于将同一种酶应用于不同反应体系,缩短研发周期和成本。与此相反,高底物杂泛性的酶可能会在复杂的体内环境中产生副反应,从而消耗能量、辅酶、关键前体等细胞资源,或者导致毒性物质的积累,因此构建细胞合成代谢途径需要优先化选择专一性较高的酶[5,39]。目前,研究者已开发了一系列的算法用来预测酶的底物杂泛性,例如:基于酶学分类对新型化学结构和反应预测的BNICE[40],基于理化性、单肽/二肽分布、分子量、等电点、氨基酸序列信息的SVM[41],基于分子电性参数、立体参数、疏水参数、取代基等参数的QSAR[42]和基于蛋白质三维结构信息的BioGPS[43]等算法。目前,未见系统性研究比较各类预测算法的效率、准确度等性能;未来酶蛋白的规模化挖掘实验可以为算法评估提供数据支持。
构建生物合成途径时还应考虑其他因素。例如,大部分算法都倾向于选择最少反应步骤的代谢途径,以减少参与酶的数量;通过预测每一步催化反应的ΔG来计算构建整条合成途径的热力学可行性;避免毒性中间产物的产生以及维持代谢流的平衡等[34]。目前,研究者已经开发出多种算法用于构建合成途径中酶的优先化选择。例如,GEM-Path可以预测在不同溶氧和生长条件下目的产物的产量[44];RetroPath则是将合成途径中每一步催化反应所需候选酶的数量和酶的底物杂泛性等因素作为计算参数,从而指导每一步催化反应所需酶的挖掘[45];Genomatica等使用SimPheny Biopathway Predictor预测得到了10000个不同的、包含4~6个基因的1,4-丁二醇合成途径,并结合酶催化反应步骤数、热力学可行性和产量等标准进行优先化排序,缩减实验量,最终得到了具有工业化价值的菌株[35]。
2 酶蛋白挖掘的高通量实验技术
由于无法从氨基酸序列准确预测酶的催化功能,需要对大量候选序列进行实验表征,筛选出与目标性能最为接近的酶元件。另一方面,由于候选酶蛋白往往来自于不同物种,需要系统性优化密码子、调控元件、细胞培养条件等实验方案,提高酶蛋白与异源底盘的适配性。为此,研究者开发了一系列高通量技术用于酶蛋白的规模化表征,以及实验方案的组合式优化[2,4]。下面分别就分子克隆、蛋白质表达和功能筛选等三个环节进行介绍。
2.1 高通量分子克隆
高效可靠的DNA组装方法对于实现候选酶元件的规模化克隆至关重要,并且可以针对表达载体、调控元件、融合蛋白等因素进行组合式优化,提高异源表达的成功率[46]。下面将介绍满足根据不同实验需要的高通量分子克隆方法(表2)。针对酶元件DNA序列的优化策略,如密码子优化、酶切位点移除等,已有文献进行系统性总结[47],本文不再复述。
2.1.1 限制性内切酶克隆系统(restriction enzymebased cloning)
近几十年,限制性内切酶克隆方法已广泛地应用于分子生物学实验当中,包括BioBrick、Flexi、Golden Gate等克隆方法。在使用常规的限制性内切酶克隆方法时,插入片段和目标载体都需要经过限制性内切酶处理,操作时需要避免目的基因内部有使用的内切酶识别位点序列,因此较难实现多基因的平行操作。稀有限制性内切酶SgfⅠ和PmeⅠ(SgfⅠ:GCGATCGC;PmeⅠ:GTTTAAAC)的发现及Flexi克隆系统的开发,使内切酶切系统具有了高通量应用的可行性。SgfⅠ和PmeⅠ识别位点序列在许多模式生物中出现频率很低(人1.2%,鼠1.2%,酿酒酵母2.96%,拟南芥2.4%,大肠杆菌6.35%),而且该方法可以将基因序列在不同的Flexi载体之间实现简单转换,不需要重新测序[48]。Nagase等[49]使用Flexi系统实现了人类基因组中的1929个基因的高通量克隆,证明了该系统在操作中的良好应用性。

表2 基因高通量克隆、组装方法Tab.2 Methods for high-throughput gene cloning and assembly
Golden Gate组装方法基于Ⅱs型限制内切酶在同一反应体系中进行酶切和质粒组装[48]。Ⅱs型限制内切酶,如BsaⅠ、BsmBⅠ等,在其识别序列的外侧进行切割产生4bp的黏性末端。因此,只需在相邻片段上合理地设计4bp的互补序列,就可进行无痕组装。理论上,4bp的序列可以组成256种不同互补区,而且不依赖于相邻片段之间的同源性,因此可实现多片段快速组装,且不受重复序列的影响;并且,通过多种限制性内切酶的替换使用,可以实现标准化基因元件的多轮逐级组装(如调控和催化元件、单一酶蛋白表达框、代谢通路等)。值得注意的是,4bp接口序列及其组合对于组装的效率、准确性等有很大影响[50],需要利用经验和算法优化接口序列设计(如http://cuba.genomefound‑ry.org/home)。基于这一组装方法,赵惠民团队[51]开发了类转录激活因子感受器核酸酶(transcrip‑tion activator-like effector nucleases,TALEN)的自动化组装方法,可以一步组装15个DNA片段,基于机器人平台每天可以合成超过400个TALEN蛋白对,成功率超过96%。此外,研究者已开发了多个基于Golden Gate的自动化DNA组装方法[52-54]。与Flexi方法类似,该组装方法的一个限制因素是需要移除目的基因序列的BsaⅠ、BsmBⅠ等酶切位点。相较于BsaⅠ或BsmBⅠ,SapⅠ的识别位点序列出现的频率更低,具有更好的通用性。此外,赵国屏课题组[55]在2013年建立了一种新型Golden Gate组装方法,称为MASTER连接法(methylation-assisted tailorable ends rational ligation method)。该方法使用同时具有Ⅱm型和Ⅱs特性的内切酶MspJⅠ,MspJⅠ只能识别甲基化的4bp位点,mCNNR(R=A或G),并在识别位点外侧进行切割,因此不受到目的序列是否包括MspJⅠ酶切位点的限制。
2.1.2 同源重组克隆系统(sequence homology-based cloning)
同源重组克隆组装依赖于载体和连接片段两端的同源序列。该方法简便高效,既可用于单片段的克隆,也可用于多片段与载体的组装,且不受到酶切位点的限制。该系统主要包含Gibson、Gateway、Echo Cloning、Creator等体外酶法组装[48],以及利用酵母高效同源重组机制的DNA assembler等胞内组装方法[56]。其中,Gateway方法在高通量克隆组装实验中应用最为广泛,该方法利用λ噬菌体与大肠杆菌的染色体之间发生的位点特异性的重组整合[attB-attP(BP反应)→attL-attR(LR反应)],克隆效率可达95%以上。其最主要优势是完成入门克隆之后,目的基因序列可以通过LR反应高效、简便地连接至其他目的载体[57]。但是,当目的基因序列的长度超过3000bp时,连接效率会降低。另外,研究者也基于Gibson assembly[52-53]、DNA assembler[58-59]等技术开发了高通量的DNA组装方法。这一类克隆系统的共同局限,是在组装含有重复性序列的片段时,容易由于序列内部的非特异性同源重组导致组装失败。
2.1.3 基于寡核苷酸的架桥法克隆系统(bridging oligo)
De Kok等[60]基于连接酶循环反应(ligase cycling reaction,LCR)开发了新型的DNA序列组装方法。通过设计与相邻DNA的两端序列互补的单链桥接寡核苷酸,在较低温度下进行退火,从而使上游片段的3'端与下游片段的5'端连接,将2个DNA片段组装成单个的线性片段。在接下来的循环中,以组装好的线性片段为模板来组装互补链,通过多次热循环,可以将线性DNA片段组装成环形质粒,并转化大肠杆菌感受态进行扩增。De Kok等利用该方法成功地将12个DNA片段连接至20kb的载体上[60]。
2.1.4基于CRISPR的克隆系统
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)是存在于原核生物中的获得性免疫系统。Cpf1蛋白隶属于Ⅱ类Ⅴ型CRISPR系统;相较于Cas9蛋白,Cpf1具有相似的基因编辑效率、蛋白质更小、具有较低的脱靶效应和较好的可操作性。由单个crRNA(CRISPR RNA)引导,Cpf1结合在富含胸腺嘧啶的PAM(photospacer adjacent motif)位点相邻并与crRNA互补的DNA序列,切割靶标DNA互补链的23位和非互补链的18位,从而形成5 nt黏性末端。Cpf1的切割位点受crRNA中靶向spacer序列长度的影响:当spacer序列长度大于等于20bp时,Cpf1倾向于切割非互补链的18位;spacer序列长度小于20bp时,Cpf1倾向于切割非互补链的14位,从而形成8 nt的长黏性末端。基于这一特性,研究者开发了CCTL(Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation)方法用于大DNA片段的体外编辑。利用17 nt长度的crRNA spacer,研究人员成功将放线菌紫红素合成基因簇actⅡ-orf4基因的启动子进行了原位替换,效率达到70%以上[61]。进一步,研究者对Cpf1进行了改造,使其可以识别60种不同的PAM位点,将其靶向范围扩大了4倍,大大增加了Cpf1可以靶向的序列范围[62]。
2.2 高通量蛋白表达
酶蛋白表达是一个复杂、高成本和耗时的过程。蛋白质表达的底盘系统包括原核细胞(如大肠杆菌和枯草芽孢杆菌)、真核细胞(如酵母、昆虫细胞和哺乳动物细胞)以及无细胞(cell-free)蛋白合成系统。本文重点介绍大肠杆菌表达系统和无细胞表达系统。
大肠杆菌表达系统是经典的重组蛋白表达系统,具有操作简单快捷、培养周期短、成本低廉、遗传背景清楚等优势,使其成为研究者首选的蛋白表达系统[63]。传统的大肠杆菌蛋白表达、纯化和检测的实验操作都由研究人员亲自动手完成,不仅烦琐,还耗费大量的等待时间。随着自动化技术成本的降低以及智能程度的提高,采用自动化机器替代人工,使得大肠杆菌蛋白表达过程可高通量、自动化地进行,而且操作更精准、更标准化。目前,国内外已搭建起若干个全自动高通量筛选平台。例如,德国格赖夫斯瓦尔德大学拥有一套全自动化高通量筛选平台用于大规模的蛋白质工程筛选。它以96微孔板等作为实验载体,通过自动化操作系统Momentum和Agilent VWorks 9软件对实验过程进行编程,自动化进行重组质粒转化、细胞培养、蛋白质表达与纯化以及蛋白质性质表征等[64]。
此外,无细胞蛋白质合成体系逐渐发展为一项快速、高效的体外合成蛋白质的技术手段。该技术是以外源的DNA或mRNA为模板,通过补充底物和能量物质,在细胞抽提物提供的酶系作用下完成蛋白质的体外表达[65-66]。细胞抽提物可以来源于大肠杆菌、兔网织红细胞、昆虫细胞和哺乳动物细胞等。该系统能够以PCR产物作为线性模板,因此无需烦琐的克隆、连接、转化、细胞裂解和蛋白质提取等步骤。且由于该系统不存在活细胞,可用来表达在胞内系统中难于表达的毒性蛋白质。传统的胞内蛋白质表达系统,从基因克隆、质粒转化、宿主菌培养、目标蛋白质表达和蛋白质分离纯化,一般需要2~3周左右的时间。无细胞蛋白质合成体系,只需要简单的基因扩增、无细胞蛋白质表达以及简单的分离纯化等步骤,整个过程一般只需要3~4天,表达周期大大缩短并节约相应的经济成本[67]。例如,Nakano等[68]开发了一种完全在体外进行的蛋白突变体库构建与筛选的高通量方法。利用无细胞蛋白质合成体系、PCR体外扩增、微孔板反应并结合ELISA筛选,对抗人血清蛋白的单链可变片段进行了体外筛选,成功筛选到具有抗体结合功能的蛋白质片段。商业化的96孔和384孔板是最为常用的较高通量的实验载体,能方便地使用与之配套的酶标仪等仪器进行检测。然而,由于受到微孔板孔数的限制,商业化微孔板中的无细胞蛋白表达难以满足通量更大的研究,因此,Angenendt等[69]定制了一种孔最大容量为1.5μL的1536孔规格的微孔板。进一步,利用绿色荧光蛋白与β-半乳糖苷酶作为检测蛋白,研究者成功将微孔板中无细胞反应体系缩小到100 nL,而且反应体系的浓度稀释10倍后反应依然能进行。
表达不同来源的酶蛋白时,通常需要针对DNA/氨基酸序列、助溶融合标签、调控元件强度、细胞培养条件等进行系统性、组合式优化;因此,需要开发高通量方法评估不同实验条件下酶的可溶性表达结果。聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)和蛋白质印迹法(Western blot)是检测蛋白质是否可溶性表达的常规方法;但其过程耗时烦琐,很难进行高通量操作。Split GFP技术提供了一种操作简便、易于自动化的可溶性表达检测方法:将GFP11片段(约15个氨基酸)融合表达在目标蛋白质的氮端或者碳端;改造后的目标蛋白质与包含GFP其余序列的重组片段(GFP1~10,约200个氨基酸)在体内或体外环境相遇时,可以结合产生绿色荧光,且荧光强度与目标蛋白质的可溶表达量呈正相关[70-71]。这一技术为快速筛选不同实验设计方案,从而优化酶元件在底盘体系中的可溶性表达提供了基础。
2.3 酶催化功能的高通量表征
2.3.1 酶反应分析方法
酶促反应动力学(enzyme kinetics)分析是对酶蛋白进行功能表征的核心手段。光谱、质谱和电化学方法是高通量酶动力学分析的常用方法。对于少数反应物具有特征光学性质的酶促反应而言,可以通过监测吸光度、荧光等信号跟踪反应进程。然而,大多数生化反应的底物或产物不产生特征的光学信号,需要通过设计酶反应分析方法(enzyme assay),如利用偶联反应、生物传感器等手段将反应物的浓度信息转化成仪器可检测的光、电等信号。例如,琥珀酸辅酶A合成过程中伴随着ATP的水解反应,形成ADP和正磷酸盐,研究者可以利用钼酸与正磷酸盐生成蓝色颜料来定量琥珀酸辅酶A合成酶的效率[72]。另一方面,生物传感器可以利用转录因子(transcriptional factor,TF)、工程荧光蛋白、核糖核酸适配子(RNA aptamer)等功能元件识别细胞内特定代谢物,并进一步通过基因调控将目标分子的浓度信息定量转化为报告基因的表达量[73-76]。
电化学传感器可检测电极表面生化反应导致的电流变化,具有很高的检测灵敏度和选择性,并且可进行微型化处理。该类型的传感器主要由生物识别元件以及可将生物信号转换成电化学信号的传感器组成。研究者开发了具有96个丝网印刷电极(96 screen-printed electrodes)的系统,用于分析半乳糖氧化酶产生的H2O2,对酶反应的检测更加灵敏、快速[77]。电极材料是影响传感器的主要因素,根据电极材料的不同分为电流计、电位计、电导计和阻抗计。目前新型的电化学传感器采用纳米材料,如碳纳米管、金属和金属氧化物纳米颗粒、硅纳米颗粒和半导体材料纳米颗粒等[78]。
与光学、电化学等分析方法不同,质谱(mass spectrometry,MS)基于离子的质荷比(m/z),可以对反应物进行无标记(label-free)的定性与定量测定,对于酶反应分析具有更好的普适性。但是,检测样品进入质谱之前需要经过耗时的色谱分离,限制了质谱筛选的通量。目前,基于新型质谱仪器设计,通过激光、微流控或声学技术将分析物直接引入质谱仪,可以在几秒钟内完成单个样品的分析。例如,基质辅助激光解析解离质谱(ma‑trix-assisted laser desorption/ionization MS,MALDI MS)可以利用激光取样,快速分析靶板表面的酶反应阵列,具有样品制备简单、高耐盐以及广泛的生物分子覆盖性等特点[79]。de Rond等[80]基于MALDI MS开发了PECAN方法,完成了P450BM3突变库的高通量筛选鉴定。电喷雾质谱(electro‑spray ionization MS,ESI MS)与微流控液滴系统联用可以进行高通量的分析与分选,在小分子分析方面相较MALDI更具优势,该技术已经应用于蛋白质工程、药物开发和诊断等研究工作[81]。另外,基于质谱高分辨率的特点,可以开发代谢组学方法,同时对数百种代谢物进行分析[82]。基于这一理论,将候选酶与细胞粗提物共同孵育,通过监测反应前后的质谱信号变化,可从大量代谢物中快速鉴定反应底物与产物;进一步结合化学计量学和数据库分析,实现酶的高通量筛选与功能鉴定[83]。
2.3.2 高通量样品处理与分析技术
基于以上酶反应分析手段,可以利用自动化移液、流式分选和微流控液滴等技术提高酶功能表征的通量和准确性。
自动化移液工作站通常由一个工作台面、移液机械臂、抓手机械臂、相关功能模块和配套电脑构成。常规的移液工作站有8通道或96通道的移液机械臂,可以实现96孔板以及384孔板的移液操作。通过合理的程序设定,可实现短时间内全自动处理大规模液体生物样本,有效提高实验的准确性、稳定性和效率。此外,在工艺流程中整合菌落挑取仪、酶标仪等功能模块,可以赋予移液工作站更加丰富、个性化的用途。例如,前述德国格赖夫斯瓦尔德大学平台通过整合移液工作站和酶标仪,发展了高通量酶活检测方法,实现了单加氧酶、转氨酶、脱卤素酶和酰基转移酶等文库大规模筛选[84]。
细胞为酶促反应提供天然环境,并将酶蛋白与其编码基因进行物理偶联;结合流式细胞荧光分选(fluorescence-activated cell sorting,FACS)以及二代测序技术,可以快速建立酶序列-功能间的对应关系。对于细胞内代谢物或具有渗透性的胞外产物,可以将酶反应与荧光蛋白的表达、折叠或运输过程偶联[85];针对非渗透性底物,可以利用表面展示技术将酶蛋白表达在细胞表面,并利用分子互作将反应物精巧固定于细胞表面,从而基于荧光探针和反应物的结合强度进行酶活筛选[86]。
对于胞外游离产物,无法利用传统FACS技术进行分析,需要结合微流控液滴技术对单细胞及其周边微环境进行分析[87]。每个细胞被分装在微流体元器件中以每秒数千滴的速度产生的独立水油小液滴中,其体积大小由通道尺寸和流体流速控制,通常从纳升到皮升不等[88]。集成微流体分选仪可在103Hz频率下筛选高荧光液滴,并根据用户定义的分选标准施加电场,将包含单细胞的液滴转移到收集室或废物室中。微流控芯片具有高灵敏度、定量读出和高准确性等优点,通过液滴注射或液滴融合可以实现多步骤酶催化反应[85]。结合先进的荧光标记分选技术、拉曼光谱和质谱等,可以显著提高基于液滴的微流体系统的筛选效率。此外,微流控技术也可应用于宏基因组中催化元件的高通量挖掘[89]。
3 高通量酶元件挖掘在酶家族研究中的应用
酶家族(enzyme family)是由序列、结构、功能、进化方面具有相似性的蛋白质组成的集合。随着高通量(宏)基因组测序技术的发展,数据库中蛋白质序列的数量呈指数增长,对功能注释和预测提出了重大挑战;而对酶家族特征信息的提取,有助于充分利用已有实验数据对未知序列的功能进行归属[90]。利用合成生物学手段系统化探索酶家族的序列-功能关系,是规模化挖掘新型酶元件的一个重要途径。
综合利用聚类分析和实验验证优先化算法,有利于提高对酶家族进行高通量挖掘的效率。卤代烷烃脱卤酶(haloalkane dehalogenases,HLD)采用水解型脱卤机制催化卤代有机化合物的碳-卤键的断裂,在环境污染物降解、手性化合物合成、分子成像等方面具有应用潜力。利用Position-Specific Iterated(PSI)-BLAST比对,Vanacek等从NCBI核酸测序数据库中识别出5661个可能的HLD[18]。为了从数千个候选序列中快速筛选出最为相关的HLD,研究者首先采用了类似SSN的成对同源比对聚类方法,初步筛选出953个较为可信的HLD序列。接着,基于多序列比对去除了117个不完整序列和178个氨基酸水平的同义序列。对剩余的658个HLD序列,研究者制定了优先化原则——序列多样性、物种来源/生境多样性、活性位点结构多样性、与已知HLD低同源性和高可溶性等,综合利用同源结构建模、可溶性预测等生信工具,研究者选择了20个HLD蛋白进行实验表征,并最终实现了8种HLD在大肠杆菌底盘中的可溶性表达(图2)。结果显示,这8种HLD来源于不同的细菌、真核生物和古菌,表现出非常多样化的底物范围和特异性、最适温度范围(20~70°C)和最适pH范围(5.7~10),并且发现了迄今催化活性最高的一个HLD蛋白[18]。这项研究表明,综合利用聚类和优先化算法,有利于从酶家族中快速挖掘具有新颖功能和性能的酶元件。
许多酶家族成员虽然具有相似功能,但在序列和结构水平上具有多样性[91]。例如,胞质谷胱甘肽转移酶(cytosolic glutathione transferase,cytGST)在新陈代谢和防御氧化损伤中具有关键作用;其超家族包含13000多个非冗余序列,催化多种反应类型(具有超过140个Enzyme commission编号),但催化机理并不完全清楚。为了系统性研究cytGST酶家族,Mashiyama等[92]首先利用Cd-hit算法[93]将数据库中的13493个cytGST按照50%的序列同一性(50% sequence identity,ID50)进行聚类,提取出2190个具有代表性的序列;针对这2190个ID50节点,研究者对衡量序列相似性的E-value进行了步进优化,并最终选定1×10-25的E-value阈值构建SSN网络,包含约30个主要的簇和超过60个较小的簇或单个节点,其中有23个簇中至少有一个成员被实验证实有GST活性。在聚类分析的指导下,研究者从不同的簇或单个节点中优先选择了857个候选基因进行实验验证。高通量分子克隆、蛋白质表达与纯化、结晶等实验在纽约SGX结构基因组学研究中心(New York SGX Research Center for Structural Genomics)开展;利用自动化移液工作站等高通量手段,该研究中心可以每月制备120个以上的超纯蛋白质样品,每年解析100~200个蛋白质晶体结构[94]。最后,共有230个候选序列在大肠杆菌中被成功表达纯化,其中27个蛋白质的37个3D晶体结构得到解析。通过筛选175个不同的底物,研究者发现了82个新的具有GST活性的酶,将具有GST活性的簇从原来已知的23个增加至35个,并发现少数成员具有非常罕见的还原性脱卤活性。研究者将新发现的蛋白质与文献报道已知具有GST活性的174个蛋白质综合分析,将其序列、结构、催化机理等信息与SSN网络的结构进行映射,用以生成序列-结构-功能关系的全局视图。结果表明,53%的cytGST具有高度底物专一性,只与1个底物发生反应;而7%的酶可以催化至少6个底物的转化。作者还发现,如果通过催化机理而非序列或结构相似性建立相关网络,可以将cytGST家族已知15个催化功能中的14个联系起来;这一观察也再次印证了仅靠序列或结构同源性无法准确预测功能相似性的现象。这一研究是利用合成生物学方法规模化挖掘酶蛋白的经典案例,证明了采取多层次聚类分析的必要性,以及酶挖掘过程中高通量实验验证的不可替代性[92]。
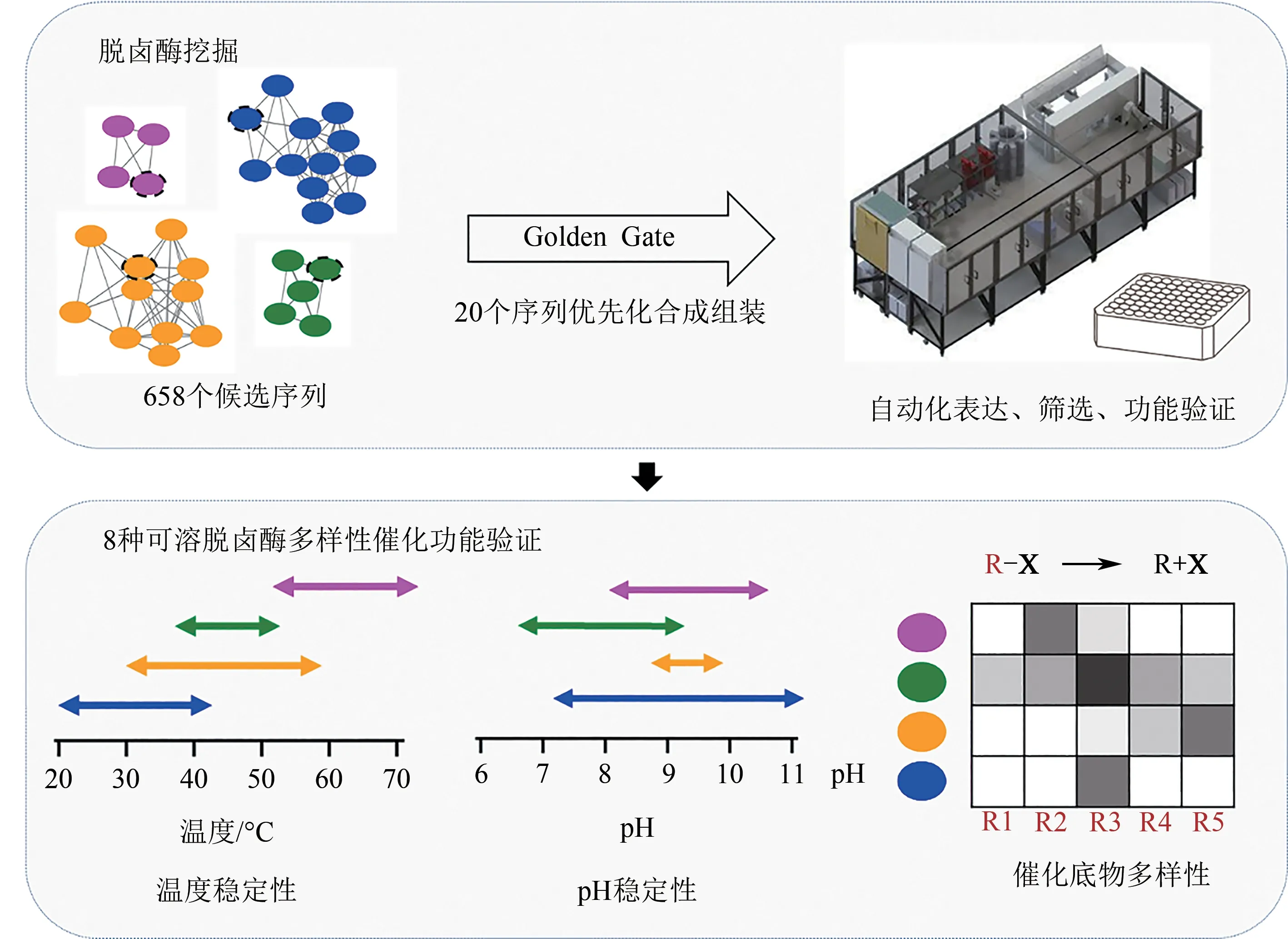
图2 脱卤酶挖掘、优先化及多样性催化功能表征Fig.2 Sequence analysis,prioritization and characterization for haloalkane dehalogenases(HLDs)
4 展望
近年来,数据库中蛋白序列的指数性增长以及生命科学前沿技术的快速发展,为人们提供了丰富的生物资源。如何充分利用全球共享的生物资源数据,对数据库中的酶资源进行高通量的挖掘利用,是研究者面临的重大机遇和挑战。综合上文所述,研究者开发了一系列算法、数据库、高通量实验技术,应用于酶蛋白的高通量挖掘,有效地推动了酶制剂和细胞工厂在生物制造方面的应用[95]。
但是,目前仍然缺乏高度集成的工程化研究平台,应用合成生物学理念和技术对酶蛋白资源进行系统化的研究和挖掘。近年来,在全球范围内已建成或在建多个大型合成生物学研究基础设施,将自动化技术应用在合成生物学“设计—构建—测试—学习”的各个研究环节。这些基础设施包括美国伊利诺伊大学的iBioFAB(Illinois Bio‑logical Foundry for Advanced Biomanufacturing)、美国能源部的Agile Biofoundry、英国爱丁堡大学的Edinburgh Genome Foundry(EGF)等。2019年,8个国家的16个隶属于公共研究机构的合成生物设施于2019年成立了“全球合成生物设施联盟”(Global Biofoundry Alliance,GBA)[96],旨在加强设施间的协作沟通,将智能制造的理念引入合成生物学。
依托这些设施平台,研究人员开发了不同程度的自动化流程进行酶蛋白挖掘。例如,本文作者在伊利诺伊大学赵惠民教授团队参与设计、搭建、运行了学术界首个全自动合成生物设施iBioFAB[97],开发了基于Golden Gate assembly的自动化DNA组装方法[51],实现了大肠杆菌和酿酒酵母的自动化转化、培养、筛选等操作[98],建立了自动化样品前处理流程对代谢物进行快速质谱表征[99],从而对酶蛋白、代谢通路、细胞工厂等合成生物体系的高通量工程构建与优化;中国科学院天津工业生物研究所团队基于自动化平台,实现了谷氨酸棒状杆菌的高通量基因编辑[100];如前所述,德国格赖夫斯瓦尔德大学开发了酶突变库自动化构建和筛选的流程,实现了单加氧酶、转氨酶、脱卤素酶等酶蛋白的定向进化研究[85]。另外,中国科学院深圳先进技术研究院正在牵头建设“深圳合成生物研究重大科技基础设施”,建成后将是我国首个将软件控制、硬件设备和合成生物学应用进行整合的大型规模化合成生物制造系统,作为开放共享平台服务我国合成生物学的科研与产业用户。通过实现生物设计、工程DNA合成与组装、底盘系统转化与培养、催化功能测试等环节的自动化运行,这些工程化平台可以高效集成酶蛋白高通量挖掘所需的数据库、算法、合成生物工艺和硬件仪器设备,从而实现优质酶催化元件的规模化挖掘,积累酶序列-功能关系的高质量定量数据,为实现酶蛋白的理性设计奠定基础。
在未来的研究中,基于合成生物学的酶元件挖掘研究可在药物、精细化工等高附加值分子的研究与生产等热点、难点领域发挥重要的作用。例如,参与内源性物质和药物、环境化合物等外源物质代谢的P450酶及其抑制剂的挖掘、筛选、功能鉴定[101];非天然和较难获得的化合物的从头生物合成,包括对未知催化途径酶的挖掘与改造、生物途径及化学途径的整合、底盘细胞的改造、人工合成途径与底盘细胞的适配等方面[35]。而依托工程化合成生物研究基础设施,可以计算设计并合成表征催化不同反应类型、适应不同实验条件的酶蛋白序列,从而建设包含功能特性清晰、符合组装标准的元件实体库。为了实现以上愿景,需要将生信分析与实验表征深度整合,通过界面友好的数据库、算法与网站,实现酶元件及其表征数据的查询、比对与二次计算,从而推荐已有元件或待合成表征的对象;开发自动合成生物实验技术,开展自动化高通量的元件挖掘、添加、存储和利用等操作,形成酶催化数据的信息化体系与元件共享平台。未来,相信合成生物学提供的工程化思想和能力可以大幅提高酶蛋白挖掘研究的通量与效率,从而加速生物制造理念的工业化实现。
