几种含钌化合物水解反应的理论探究
2020-10-09王章涛包德才
李 慧,王章涛,包德才,张 强
(渤海大学 化学化工学院,辽宁 锦州121013)
0 引言
目前,癌症被世界卫生组织列为世界五大疑难杂症之一,是具有高致死率的疾病,研发可以治疗癌症的药物一直是人们研究的重点.自顺铂类药物被发现具有抗恶性肿瘤活性之后,含有过渡金属的抗肿瘤药物受到人们的青睐.经历了近三十年的研究发展,科学工作者已相继成功开发了顺铂(cisplatin)[1,2]、卡铂(carboplatin)[3-5]等药物用于临床治疗恶性肿瘤.但目前,临床应用铂类抗肿瘤药物最大的问题是毒副作用和耐药性.既含铂类药物之后,含过渡金属钌的抗恶性肿瘤药物因其具有的高抗肿瘤活性和低毒副作用而被广泛研究.国际上报告的具有抗肿瘤活性的含钌配合物大致包含KP1019型配合物[6,7]、NAMI型配合物[8,9]、钌(II)芳烃配合物[10]和钌多吡啶配合物[11]四类.其中NAMI-A是第一个进入临床试验的含钌抗恶性肿瘤配合物.
从含钌配合物的抗肿瘤活性被发现至今,人们进行了大量的试验来探究其抗肿瘤机理.关于NAMIA抗肿瘤机理的研究发现,在生理环境下,含钌配合物会发生一系列的水解反应,水解过程与其抗癌活性有着密切关系.目前,关于NAMI-A进入生物体后,要先发生水解反应后再发挥其抗癌作用的理论,已经得到了广泛认可. 实验显示,在37 ℃的磷酸缓冲溶液中,NAMI-A水解反应的半衰期少于30 min,所以NAMI-A的水解稳定性是注射应用NAMI-A的一个限制因素[12].实验和理论上均有关于NAMI-A水解机理的报道[8,13-16].Sava等人研究了NAMI-A的不同水解产物与DNA之间的相互作应,并考察了水解反应对NAMI-A抗癌活性的影响[13].Bacac等人[15]利用NMR光谱的方法研究了NAMI-A分子在溶液中的稳定性和水解反应.陈等人[16]首次利用理论方法研究了NAMI-A的第一步和第二步水解机理.Vargiu等人[17]在研究NAMI-A水解的同时也探究了氧化还原性质对水解反应的影响.NAMI-A是以Ru(III)离子为中心,拥有六个配体的八面体构型的分子,其中四个氯离子配体位于八面体的横轴平面上,咪唑环(Im)和二甲亚砜(DMSO)基团位于纵轴的两端.我们在之前的研究中探究了在显性溶剂模型下NAMI-A的多步水解机理,结果显示NAMI-A中的咪唑环配体的水解反应很难发生[18].但是咪唑环配体是否会对水解反应产生影响是值得探究的问题之一,本文中,我们分别选择了吲唑配体、1,2,4-三唑配体、1-氨基-1,2,4-三唑配体和1-甲基-1,2,4-三唑配体来代替NAMI-A中的咪唑环配体,得到四种NAMI-A的类似物,利用理论方法探究了四种化合物的水解反应路径并计算了水解反应所需的活化自由能.
1 模拟细节
我们利用DFT理论[19]中的B3LYP方法[20,21],在没有任何限制性条件下进行优化得到反应过程中的所有结构,包括反应加合物(RA)、过渡态(TS)和产物加和物(PA),所有计算均是采用LanL2DZ(用于金属钌)和6-31G(d,p)(用于体系中的其他原子)混合基组. 在结构优化的同时进行了频率的计算,所有的过渡态结构中都具有一个虚频,并进一步完成了内禀反应坐标(IRC)的验证. 而得到的其他各驻点结构均无虚频. 为了考虑溶剂环境的影响,水解反应路径上的构型均是经过QM/MM(ABEEM)[18]迭代优化得到,QM区域的计算均是在MM区域作为背景点电荷的形式下完成. 迭代优化的收敛标准为两次QM区域优化后计算所得到的能量最大偏差在5×10-4a.u.范围内,MM区域优化得到的均方根偏差(RMSD)在5×10-3kcal·mol-1·A-1范围内.
根据迭代优化得到的结构,利用QM/MM(ABEEM)与自由能微扰理论相结合的方法计算反应的活化自由能.利用ABEEMσπ浮动电荷极化力场完成自由能微扰计算中的动力学模拟过程.从反应加合物到过渡态以键长为变量进行二十等分,得到反应路径上的每一个结构,再分别放入边长为18.62 Å的立方体水盒子中,加入抗衡离子.动力学模拟是在正则系综(NVT)下加入周期性边界条件和最近镜像中完成,采用Verlet跳蛙积分运动方程,积分步长设为1.0 fs.控制温度采用的是Berendsen法则,设定295 K为模拟过程中的初始温度.截断半径为9.0 Å.每一个体系的前100 ps动力学模拟过程用来使体系达到平衡,后100 ps的数据则用来统计平均并进行计算.自由能差的计算是在采用双向取样方法的基础上,计算平均值而得到.
2 结果与讨论
2.1 四种化合物的结构
我们选取含有不同含氮杂环配体的NAMI-A类似物作为研究对象,首先探究了其结构信息.将含有吲唑配体的含钌复合物称为化合物1,含有1,2,4-三唑配体的含钌复合物称为化合物2,含有1-氨基-1,2,4-三唑配体的含钌复合物称为化合物3,含有1-甲基-1,2,4-三唑配体的含钌复合物称为化合物4.优化后的结构如图1所示.
表1中给出了化合物结构中与中心金属Ru成键的化学键的键长.我们看到四种化合物中的Ru-S键长几乎相同,化合物1中的Ru-N键要长于其他三种化合物中的Ru-N键.化合物1中的Ru-Cl(1)键长大于其他三种化合物中的Ru-Cl(1)键,化合物2中的Ru-Cl(2)键和Ru-Cl(3)键均长于其他三种化合物中的Ru-Cl(2)键和Ru-Cl(3)键,化合物3中的Ru-Cl(4)键长要大于其他三种化合物中的Ru-Cl(4)键.
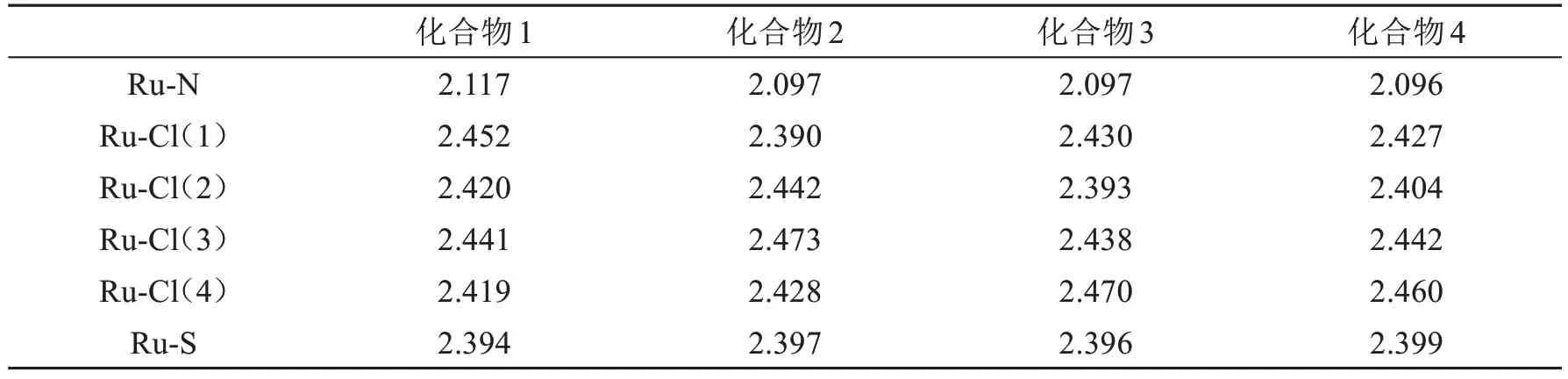
表1 四种化合物中与中心Ru成键的键长(单位:Å)
2.2 反应路径及活化自由能
我们分别研究了四种NAMI-A类似物上氯离子配体的水解反应.模拟了其反应路径上各驻点的结构并计算了反应所需的活化自由能.图2展示了化合物1中氯离子配体水解反应路径上反应加合物(RA),过渡态(TS)和产物加合物(PA)的结构及涉及化学反应的相应化学键的键长信息.从图中可以看出,在反应加合物中,进攻水分子上的氧原子与吲唑配体上的H原子间形成氢键,同时水分子上的两个H原子也会和临近的两个氯离子配体有氢键作用.而在过渡态结构中,原来的氢键网络被破坏,水分子中的两个H原子一个趋向于与离去氯离子形成氢键,另一个倾向于与DMSO基团上的O原子形成氢键,直至产物加合物中,水分子中的O原子与中心金属Ru配位后,水分子配体上的H原子分别与离去的氯离子和DMSO中O原子形成氢键.从图中标示的键长信息我们可以看出,从反应加合物到过渡态,进攻的水分子中的O离中心金属Ru的距离从4.08 Å缩短到2.48 Å,直到产物加合物中Ru-O键形成(键长为2.10 Å).同时,氯离子配体到中心金属Ru的距离从反应加合物中的2.44 Å增大到过渡态中的2.90 Å,直至到产物加合物中Ru-Cl键完全断裂,距离为4.27 Å.从键长变化中我们可以发现Cl离子配体被水分子取代.
图3展示了化合物2中氯离子配体水解反应路径上反应加合物(RA),过渡态(TS)和产物加合物(PA)的结构及涉及化学反应的相应化学键的键长信息.从图中可以看出,从反应加合物到产物加合物,进攻水分子上的氧原子与1,2,4-三唑配体上的H原子间形成的氢键消失,同时水分子上的两个H原子从与临近的两个氯离子配体形成氢键变成了分别与离去的氯离子和DMSO中O原子形成氢键.从键长变化中我们可以看出,从反应加合物到过渡态再到产物加合物,进攻的水分子中的O离中心金属Ru的距离从4.29 Å缩短到2.65 Å再到2.12 Å,Ru-O键形成.同时,氯离子配体到中心金属Ru的距离从2.46 Å增大到3.00 Å,直到Ru-Cl键完全断裂(距离为4.27 Å),Cl离子配体被水分子完全取代.
图4展示了化合物3中氯离子配体水解反应路径上反应加合物(RA),过渡态(TS)和产物加合物(PA)的结构及涉及化学反应的相应化学键的键长信息.从图中给出的键长信息我们可以看出随着进攻水分子不断靠近中心金属Ru,氯离子配体离中心Ru离子越来越远.从反应加合物到产物加合物,进攻的水分子中的O离中心金属Ru的距离从4.26 Å缩短到2.07 Å,氯离子配体到中心金属Ru的距离从2.44 Å增大到4.07 Å,取代反应发生.在产物加合物中,与中心金属Ru配位的水分子配体上的两个氢原子分别与离去氯离子和DMSO基团中的O原子形成氢键.
图5展示了化合物4中氯离子配体水解反应路径上反应加合物(RA),过渡态(TS)和产物加合物(PA)的结构及涉及化学反应的相应化学键的键长信息.从图中可以看出,随着Ru与进攻水分子中的氧原子之间距离的不断减小,进攻水分子中的两个氢原子在反应加合物中分别与临近的两个氯离子配体形成氢键,在产物加合物中分别与离去的氯离子和DMSO基团中的O原子形成氢键.反应加合物中,进攻的水分子中的O离中心金属Ru的距离为4.27 Å,Ru-Cl键长为2.39 Å,反应后,在产物加合物中,氯离子离中心金属Ru的距离增大到4.08 Å,Ru-O(H2O)形成,键长为2.39 Å.氯离子配体被水分子完全取代.

表2 四种化合物发生氯离子配体水解反应所需的活化自由能(单位:kcal/mol)
我们分别计算了四种化合物氯离子配体水解反应所需的活化自由能,结果如表2所示.对比四种含钌化合物的水解反应活化自由能可以发现,化合物1发生第一个氯离子配体水解反应所需的活化自由能最小,水解反应最易发生.而化合物4中氯离子配体被水分子取代所需的活化自由能为34.45 kcal/mol,是四种化合物中最难发生水解反应的.
3 结论
本文用吲唑配体、1,2,4-三唑配体、1-氨基-1,2,4-三唑配体和1-甲基-1,2,4-三唑配体来代替NAMIA中的咪唑环配体,优化得到了四种NAMI-A类似物的结构.对比键长信息可知,以吲唑为配体的化合物1中的Ru-N键要长于其他三种化合物.我们模拟了四种化合物的水解反应路径,得到了反应路径上反应加合物、过渡态和产物加合物的结构,并计算了反应所需的活化自由能.通过反应路径上的各驻点结构信息可知,四种化合物中氯离子配体的水解反应均可发生,得到的产物中Ru-O键长均小于原化合物中的Ru-Cl键长.对比计算得到的反应活化自由能,四种化合物氯离子配体水解反应所需的活化自由能顺序为化合物1<化合物2<化合物3<化合物4,说明化合物1中Cl离子配体的水解反应最易发生.
