联合TCGA和GEO数据库构建和分析胃癌中circRNA相关的ceRNA网络
2020-09-24廖苑君孙胜南蓝树金赵小蕾覃继恒饶绍奇
李 让,廖苑君,孙胜南,蓝树金,赵小蕾,覃继恒,饶绍奇*
(广东医科大学a.公共卫生学院;b.医学系统生物学研究所,中国广东东莞523808)
作为严重危害人类健康的消化道恶性肿瘤之一,胃癌的发病率和死亡率在恶性肿瘤中分别处于第5位和第3位[1]。各种治疗手段的应用使得胃癌患者的5年生存率有所上升,但胃癌晚期患者的预后仍然较差。目前,胃癌仍是威胁人类健康的复杂疾病,其发生、发展机制尚未完全明确。
竞争性内源RNA(competing endogenous RNA,ceRNA)的概念由哈佛大学医学院Poliseno等[2]首先提出,其并非一种全新的RNA,而是代表了一种新的基因表达调控模式。既往研究表明,微RNA(microRNA,miRNA)通过与靶mRNA的3′非翻译区(3′-untranslated region,3′-UTR)结合抑制靶mRNA的翻译,实现在转录后水平调节基因的表达,从而调控肿瘤细胞的生长、发育[3]。在ce-RNA的理论假说中,任何拥有miRNA反应元件(miRNA response element,MRE)结构的RNA都可以作为ceRNA,包括环状RNA(circular RNA,circRNA)、长链非编码 RNA(long noncoding RNA,lncRNA)、mRNA和假基因转录物等。作为ceRNA家族的一员,circRNA同样能够通过竞争性地结合miRNA来调控基因的表达[4]。circRNA因5′和3′端通过共价键相连而呈封闭环状结构[5],相对于线性RNA,特殊的结构使得circRNA拥有更强的稳定性和更高的保守性[6]。随着高通量测序技术的发展,大量潜在的circRNA被发现,并且被证实作为肿瘤的重要调节因子与癌症的发生密切相关[7~9]。近年来,越来越多的研究表明circRNA在胃癌的发生、发展中起着重要作用[10~11]。此外,circ-RNA具有的分布范围广、保守性高、组织表达特异性高等特点,也预示着其有望成为胃癌诊断的生物标志物或胃癌治疗的靶点[12~13]。
基于生物信息学分析和多数据库的联合应用,本研究挖掘了胃癌中差异表达的circRNA、miRNA和mRNA并构建了ceRNA网络;随后,利用网络的拓扑属性分析手段,鉴定了核心的circ-RNA并提取了circRNA介导的ceRNA子网;最后,以ceRNA机制为切入点研究了核心circRNA在胃癌发生发展中的作用机制,这为胃癌的诊断与治疗提供了潜在靶点,同时也为研究circRNA在胃癌中的作用机制提供了新的思路。
1 材料和方法
1.1 数据来源
从GEO(Gene Expression Omnibus)数据库[14]中按照以下标准获取胃癌的circRNA基因芯片数据:以“gastric cancer”&“circRNA”为关键词检索与胃癌相关的circRNA基因芯片;选择物种为“Homo sapiens”、研究类型为“expression profiling by array”两个过滤条件进一步筛选数据;最后选定实验设计符合病例对照研究类型并且病例与对照的组织样本数均大于或等于3的芯片。根据筛选结果,我们下载了两套胃癌的circRNA基因芯片:GSE13-1414(包含3个胃癌组织和3个非癌组织)和GSE100170(包含5个胃癌组织和5个非癌组织)。
作为目前最权威的癌症基因信息数据库,TCGA(The Cancer Genome Atlas)数据库[15]覆盖 33 种癌症类型,含有超过30 000例的肿瘤样本以及20 000个基因的表达信息。本研究利用gdc-client工具从TCGA数据库下载了胃癌的RNA-seq和miRNA-seq数据,其中RNA-seq数据包括30个正常样本和343个肿瘤样本,miRNA-seq数据包括45个正常样本、446个肿瘤样本及406个病人的生存数据。
1.2 差异表达RNA的筛选
利用 R 语言(版本 3.5.0)和 Bioconductor平台中的limma包对芯片数据进行差异分析[16],筛选差异表达的circRNA(DE-circRNA),纳入标准为:P value<0.05,|log2fold change(log2FC)|>1。取 GSE1-31414和GSE100170芯片中差异表达的circRNA的交集作为最终筛选出来的DE-circRNA。
对于从TCGA数据库下载的RNA-seq和miRNA-seq数据,首先去除平均表达值低于1的mRNA和miRNA,随后利用edgeR包进行差异分析[17],最后通过错误发现率(false discovery rate,FDR)校正,以 FDR<0.05 同时|log2FC|>1 为标准筛选出差异表达的miRNA(DE-miRNA)和mRNA(DE-mRNA)。
1.3 ceRNA网络的构建
首先,采用CircInteractome数据库预测DE-circRNA和DE-miRNA的调控关系[18],构建circ-RNA-miRNA关系对。随后,利用miRDB、miRTarBase和TargetScan数据库预测DE-miRNA和DE-mRNA的调控关系[19],将同时在任意两个或两个以上数据库都被预测到的miRNA-mRNA调控关系纳入后续的分析中。最后,基于共享的miRNA构建circRNA-miRNA-mRNA ceRNA调控网络,并利用 Cytoscape 软件(版本 3.6.1)对调控网络进行可视化[20]。
1.4 关键网络的提取
研究表明网络中的核心节点(拓扑属性前10%~20%)在疾病网络中扮演者关键角色[21~22],而这些核心节点一旦受到破坏,将会影响网络的功能,进而影响疾病的发生发展[22]。本研究利用Cytoscape软件的NetworkAnalyzer工具对构建的ceRNA网络进行拓扑属性分析[23],合并度数(degree)、中介中心性(betweenness centrality)、辐射力(radiality)3种指标,取网络中处于前10%的节点作为核心节点,并根据这些核心节点从原始的ceRNA网络中提取子网。
1.5 功能富集分析和生存分析
通过在线网站KOBAS对子网进行KEGG(kyoto encyclopedia of genes and genomes)通路富集分析[24],分析子网行使的生物学功能。整合来自TCGA数据库中胃癌的RNA表达数据和生存数据,使用R语言的survival包对子网进行生存分析,以挖掘预后相关的基因。
本研究遵循上述流程开展,技术路线见图1。
2 结果
2.1 差异表达的RNA
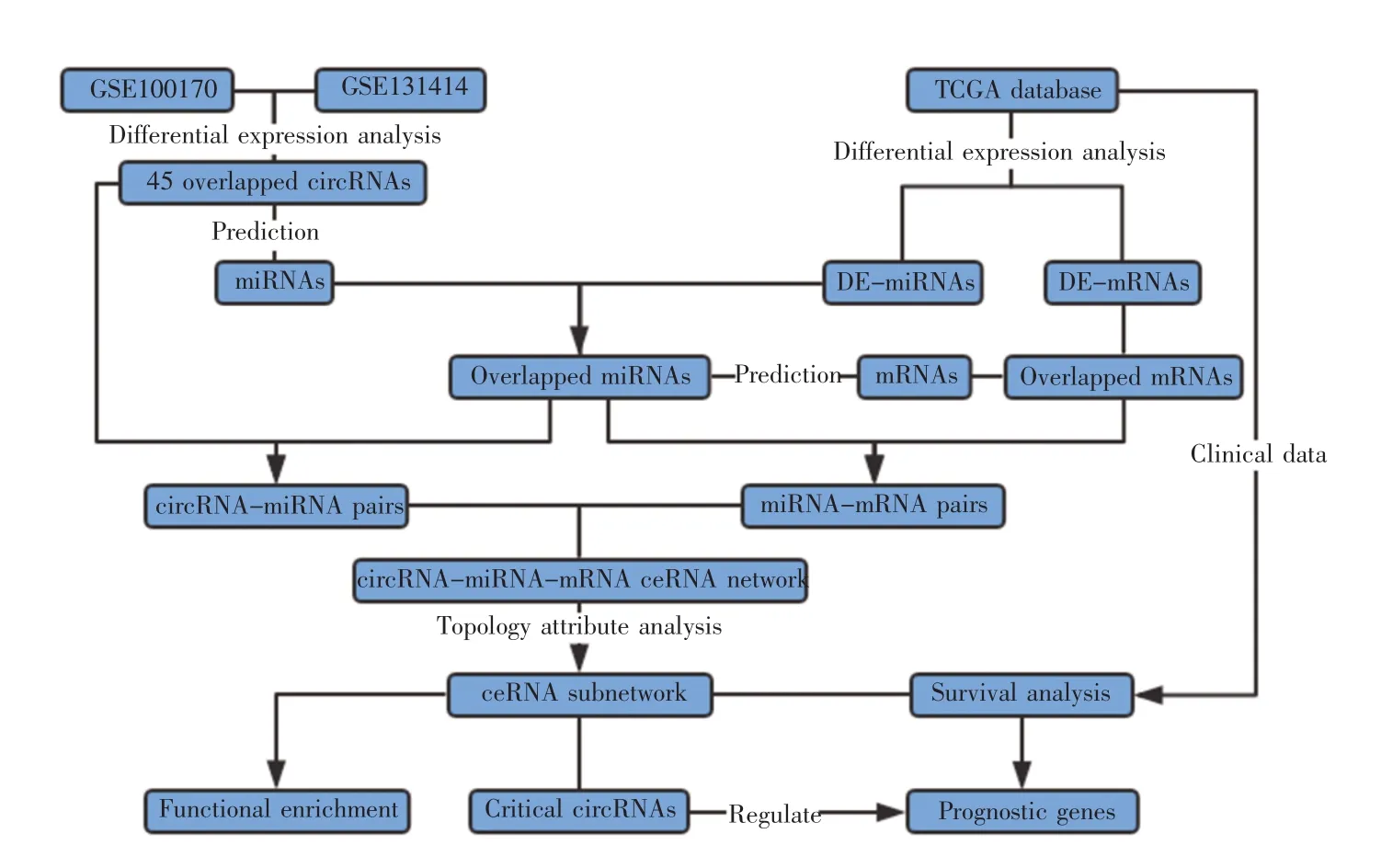
图1 数据获取及分析流程图Fig.1 The flow chart of data acquisition and analysis
芯片数据的差异表达分析结果显示,在GSE131414与GSE100170芯片中分别获得了1 522个和713个差异表达的circRNA,两者取交集后得到45个circRNA(图2A~C)。对于RNA-seq数据和miRNA-seq数据,差异分析后分别获得了4 610个差异表达的mRNA和276个差异表达的miRNA(图 2D,E)。.
2.2 原始ceRNA网络的构建
通过数据库的预测构建了1 538对circ-RNA-miRNA和2 029个miRNA-mRNA关系对。随后,基于共享miRNA构建了一个包含153对circRNA-miRNA和2 029个miRNA-mRNA关系对的ceRNA调控网络(图3),其中包含39个circ-RNA、30个miRNA和1 303个mRNA。
2.3 circRNA介导的ceRNA子网络
对原始ceRNA网络的拓扑属性进行分析后发现,3个 circRNA节点(hsa_circ_0008468、hsa_circ_0005822、hsa_circ_0025842)和 3 个 mi-RNA 节点(hsa-miR-940、hsa-miR-944、hsa-miR-515-5p)在3种拓扑学指标排名中都处于前10%,提示它们为网络的核心节点。根据3种指标对节点进行排序,前10个核心节点的拓扑参数见表1。基于鉴定到的6个核心节点,我们从原始的ceRNA网络中提取了子网络,其中包括3个circ-RNA、3个 miRNA和 501个 mRNA,共有 8对circRNA-miRNA和539个miRNA-mRNA关系对(图 4)。
2.4 子网的功能富集分析
KEGG通路富集分析结果显示,子网富集于代谢途径(hsa01100)、cAMP 信号通路(hsa04024)、神经活性配体-受体相互作用(hsa04080)、pathways in cancer(hsa05200)等多个有统计学意义的信号通路(图 5)。
2.5 子网的生存分析
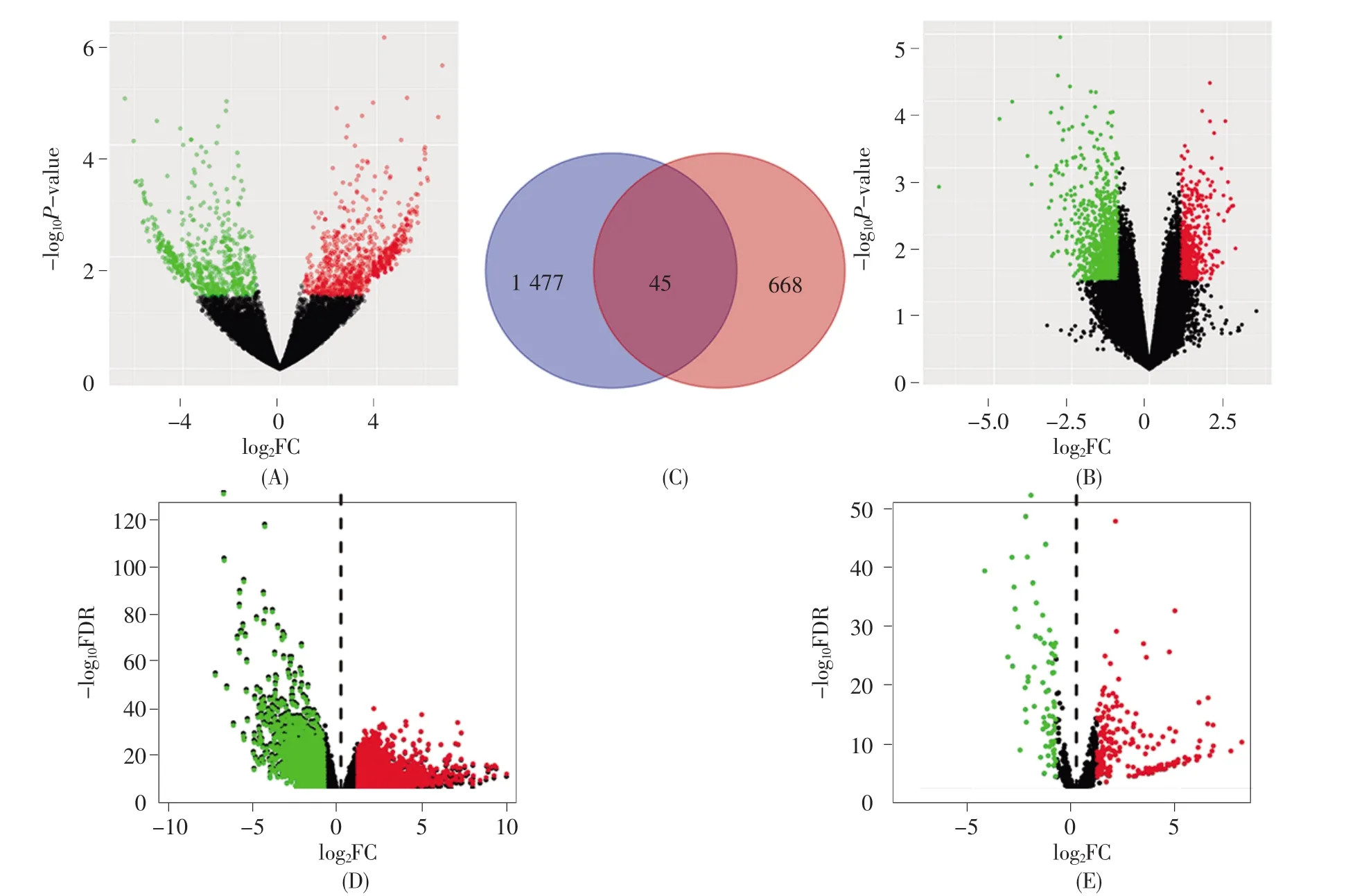
图2 胃癌中差异表达的RNA(A)GSE100170芯片中差异表达的circRNA;(B)GSE131414芯片中差异表达的circRNA;(C)芯片中差异表达circRNA的韦恩图;(D)TCGA数据库中差异表达的mRNA;(E)TCGA数据库中差异表达的miRNA。红色和绿色的点分别代表上调和下调的RNA;黑色的点代表变化不显著或变化倍数小的RNA。Fig.2 The differentially expressed RNAs in gastric cancer(A)Differentially expressed circRNAs in the GSE100170 chip;(B)Differentially expressed circRNAs in the GSE131414 chip;(C)Venn diagram of differentially expressed circRNAs;(D)Differentially expressed mRNAs in the TCGA database;(E)Differentially expressed miRNAs in the TCGA database.The red and green dots represent up-and down-regulated RNAs,respectively,and black dots represent the RNAs that do not reach the threshold for screening differentially expressed RNAs.
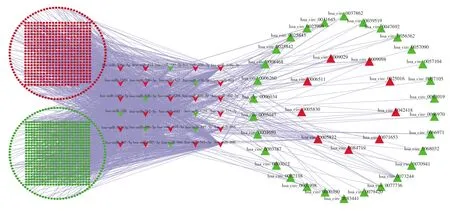
图3 胃癌中circRNA介导的ceRNA网络三角形、倒三角和圆形分别代表差异表达的circRNA、miRNA和mRNA;红色代表上调,绿色代表下调。Fig.3 The circRNA-mediated ceRNA network in gastric cancerTriangles,inverted triangles and circles represent DE-circRNA,DE-miRNA and DE-mRNA,respectively.The colors red and green represent up-regulation and down-regulation,respectively.
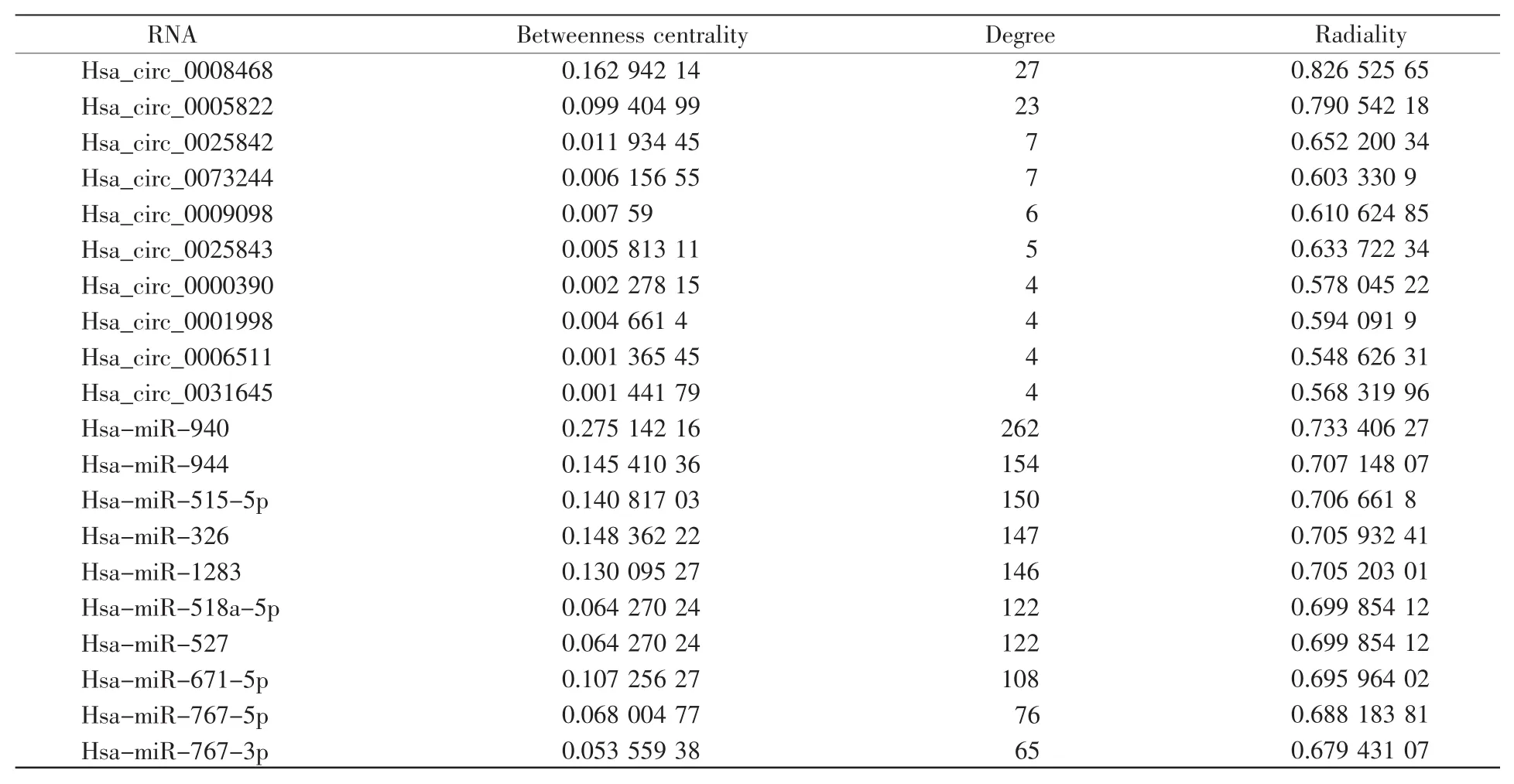
表1 原始ceRNA网络中排名前10的circRNA和miRNA的拓扑属性Table 1 Topological properties of the top 10 circRNAs and miRNAs in the primary ceRNA network
生存分析发现子网中有14个基因与胃癌的预后显著相关(P<0.01)。其中,ACO2、E2F8 的高表达组相对于低表达组预后较好;而GHR、ITIH5、KLHDC8A、NPAS3、PDE2A、PDGFD、PNMA2、RAB9B、RECK、SLC24A2、TMEM55A、TMTC1 的高表达组相对于低表达组预后较差,结果见图6。
3 讨论
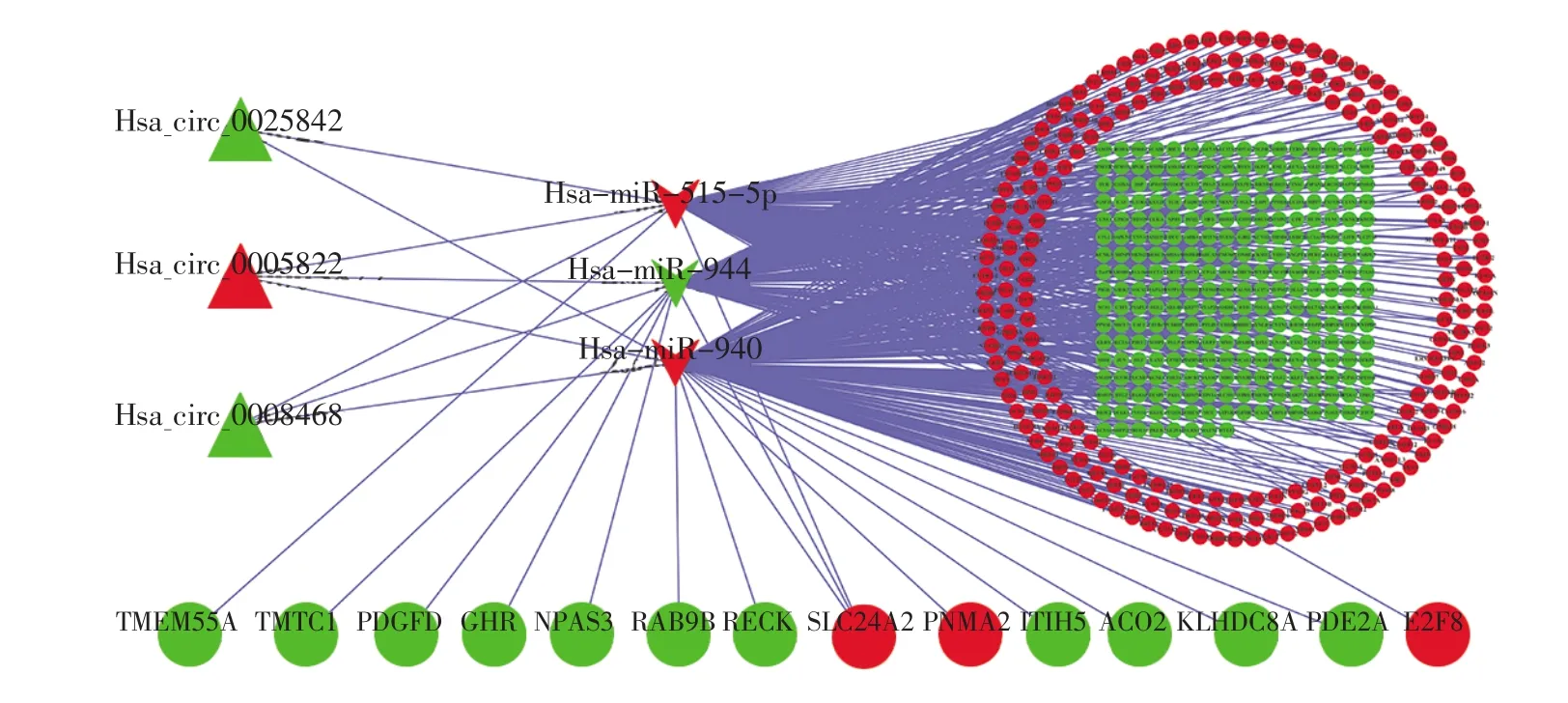
图4 竞争性内源RNA子网三角形、倒三角和圆形分别代表差异表达的circRNA、miRNA和mRNA;红色代表上调,绿色代表下调。Fig.4 The ceRNA subnetworkTriangles,inverted triangles and circles represent DE-circRNA,DE-miRNA and DE-mRNA,respectively.The colors red and green represent up-regulation and down-regulation,respectively.
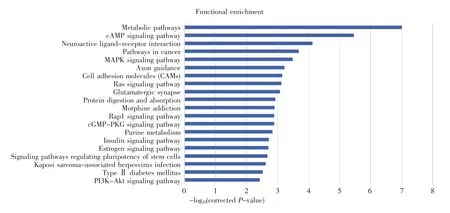
图5 KEGG功能富集图Fig.5 The KEGG functional enrichment diagram
ceRNA概念的提出将传统的相互作用模式由“miRNAs→mRNAs”网络变成更加多元的“RNAs→miRNAs→mRNAs”复杂调控网络[25]。在“miRNAs→mRNAs”互作网络中,miRNA通过以单个miRNA调节多个不同mRNA或以多个不同的miRNA调节单个mRNA的方式作用于靶基因3′-UTR区域,实现对mRNA的降解或功能抑制。ceRNA概念的提出赋予了circRNA等非编码RNA新的生物学功能[26],极大地丰富了转录调控网络。ceRNA竞争性结合miRNA,降低miRNA对靶基因mRNA的抑制作用,多种ceRNA、miRNA与mRNA之间的相互作用形成复杂的ceRNA调控网络[27],从而参与包括肿瘤发生发展在内的一系列生物学过程[28~29]。随着对circRNA认识的不断加深,circRNA被证实在肿瘤基因表达中扮演着不可或缺的调控作用[30],并且在肿瘤的发生发展中起着重要作用[31]。本研究通过GEO和TCGA数据库的联合应用构建了胃癌中由circRNA介导的原始ceRNA调控网络,并进一步通过网络分析挖掘了紧密联系的核心节点,提取了关键子网。最后,对关键子网进行了功能富集分析,以判断网络行使的生物学功能;同时,通过生存分析挖掘了与胃癌预后相关的基因。
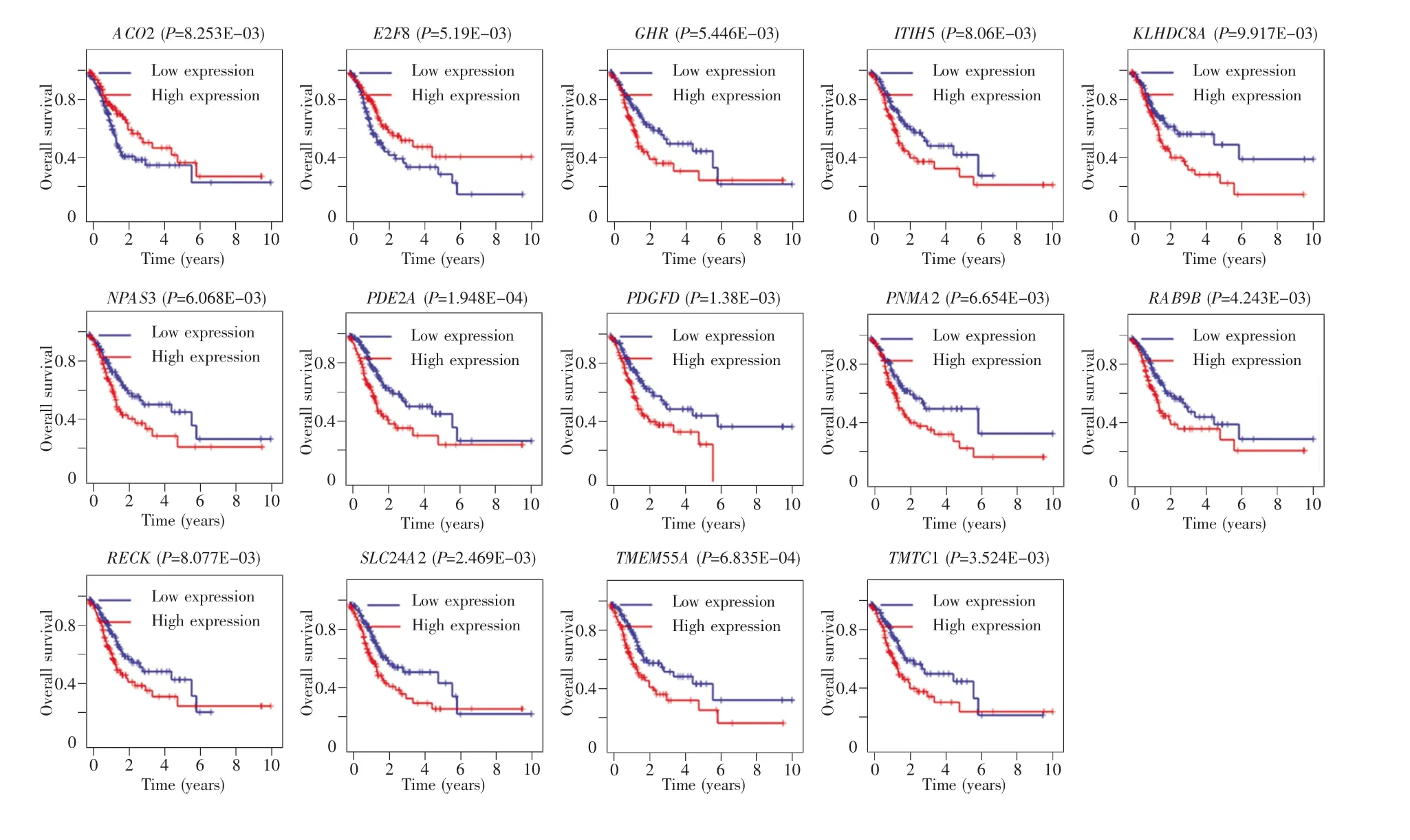
图6 ceRNA子网中14个预后基因的生存曲线Fig.6 The survival curves of 14 prognostic genes in ceRNA subnetwork
相关研究发现,miRNA通过调控靶基因表达参与细胞增殖、凋亡和侵袭等多种生物学行为[26]。利用网络拓扑属性分析,本研究挖掘了3个核心miRNA,分别为 hsa-miR-940、hsa-miR-944、hsamiR-515-5p。通过文献搜索发现,这3个核心节点已被报道与胃癌密切相关,例如:Fan等[32]研究表明,hsa-miR-940通过hsa-miR-940/Cbl-b/STAT5a轴上调PD-L1(programmed death ligand-1)的表达,从而促进胃癌细胞的增殖和迁移;Pan等[33]研究证实,hsa-miR-944通过与MACC1(metastasis-associated in colon cancer 1)的 3′-UTR 序列结合下调MACC1在胃癌细胞中的表达,从而抑制上皮-间质转化和胃癌的转移;Wang等[34]报道,hsa-miR-515-5p表达水平的上升抑制了胃癌的发生,并与胃癌细胞的生长和侵袭密切相关。文献检索的结果不仅证实了从原始ceRNA网络中鉴定的3个核心miRNA在胃癌发生发展中扮演着重要角色,同时也印证了本文分析方法在网络核心节点的识别方面拥有较高的准确性。
通过对ceRNA子网进行KEGG信号通路的富集分析,我们发现子网显著富集到代谢途径(hsa01100)、cAMP 信号通路(hsa04024)、神经活性配体-受体相互作用(hsa04080)、pathways in cancer(hsa05200)等多个有统计学意义的信号通路(图5)。其中,大部分通路已有文献支持与肿瘤发生机制密切相关,如经典的PI3K-Akt信号通路,当其被酪氨酸激酶受体激活后,磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)催化磷脂酰肌醇4,5-双磷酸磷酸化为磷脂酰肌醇3,4,5-三磷酸,导致3-磷酸肌醇依赖性蛋白激酶1(3-phosphoinositide dependent protein kinase 1,PDK1)和蛋白激酶B(protein kinase B,PKB;也称作Akt)募集到质膜并被激活,而后Akt作用于多个下游效应因子,影响肿瘤细胞的生长、增殖和凋亡[35]。另外,Yin等[36]研究表明,嘌呤核苷酸是肿瘤细胞增殖的基础和必要条件,在肿瘤细胞中嘌呤合成途径的酶代谢得到了增强,而且嘌呤代谢受损与癌症的发展有关。Boroughs等[37]研究报道,癌细胞的生长和繁殖增加了其对营养物质的摄取,而各种代谢途径可为癌细胞的生长和增殖提供必要的物质支持,其中代谢重编程被认为是癌症的标志[38]。以上信息提示,hsa_circ_0008468、hsa_circ_000-5822和hsa_circ_0025842介导的ceRNA网络参与了胃癌发生发展的多个生物学过程。
在生存分析中我们发现circRNA介导的子网中存在多个与胃癌预后相关的基因(图6)。而3个核心的circRNA(hsa_circ_0008468、hsa_circ_000-5822、hsa_circ_0025842)可能通过 circRNA-mi-RNA-mRNA调控轴调节这些预后基因的表达,从而影响胃癌病人的预后。例如:在hsa_circ_002-5842-hsa-miR-940-ACO2调控轴中,hsa_circ_002-5842 下调、hsa-miR-940 上调、ACO2 下调(图 4)。hsa_circ_0025842的低表达会减弱其对hsa-miR-940的竞争性吸附,使得过量的hsa-miR-940与ACO2结合,从而使ACO2表达水平降低。本研究的生存分析结果(图6)以及相关研究[39]都表明,ACO2低表达的胃癌患者相对于ACO2高表达的患者拥有较低的生存率。因此,我们推测hsa_circ_0025842的低表达可能与胃癌病人的不良预后相关。又如:在hsa_circ_0005822-hsa-miR-944-SLC24A2调控轴中,hsa_circ_0005822上调、hsamiR-944下调、SLC24A2上调(图4)。这说明hsa_circ_0005822的上调可促进SLC24A2的表达,而本文的生存分析结果显示SLC24A2高表达的胃癌患者拥有较低生存率(图6),提示hsa_circ_000-5822的高表达与胃癌患者不良预后相关。另外,在hsa_circ_0008468-hsa-miR-940-PDE2A调控轴中,hsa_circ_0008468下调、hsa-miR-940上调、PDE2A下调(图4),提示hsa_circ_0008468的下调抑制了PDE2A的表达。相关研究发现PDE2A的低表达会抑制肿瘤细胞的生长与侵袭[40],同时本研究的生存分析表明PDE2A低表达的胃癌患者拥有更佳的预后。因此,我们推测hsa_circ_0008468的低表达可改善胃癌患者的预后。综上所述,在ceRNA网络中,3个核心的circRNA可能通过竞争性地结合miRNA实现对预后基因的调控,从而影响胃癌患者的预后。
总的来讲,本研究通过生物信息学的方法挖掘了原始ceRNA网络中核心的circRNA,并提取了核心circRNA介导的ceRNA子网,为circRNA的ceRNA机制研究提供了参考方向,同时也为胃癌的诊断、治疗提供了新的分子靶点。研究过程采用的方法具有普适性和创新性,为后续深入的实验性研究提供了基础。
