漆酚酯键合硅胶液相色谱固定相的制备与应用
2020-09-23姚兴东李国祥雷福厚史伯安
曾 磊, 曹 宇, 姚兴东, 李国祥, 雷福厚*, 史伯安*
(1. 湖北民族大学化学与环境工程学院, 湖北 恩施 445000;2. 广西民族大学广西林产化学与工程重点实验室, 广西 南宁 530006)
高效液相色谱作为一种高效的分析方法,被广泛用于生物医学、食品安全、环境监测、生命科学和其他分析领域[1]。作为色谱分离的核心部分,色谱固定相材料的制备和应用已成为色谱研究的前沿和热点[2-5]。多孔硅胶基质由于其较大的表面积、机械强度高、稳定性好、制备简单、粒径可控等优点,常用作色谱柱固定相[6-8]。为了满足不同的分离需求,色谱研究者通常用硅烷偶联剂对硅胶进行硅烷化改性,再将设计的色谱配体键合到硅烷化硅胶上以得到色谱固定相[9-12],从而制备一系列具有不同分离性能的色谱柱。
研究表明,天然化合物独特的结构使其具有作为色谱配体的能力,能用于相似结构物质的分离富集,并且由于其具有天然、低毒或无毒性质,人工合成配体无法达到这种效果[13-15]。生漆是我国特有的优质天然植物资源,对环境友好,可生物降解。漆酚是生漆的主要成分,是一种具有15或17个碳原子的烷基长侧链的邻苯二酚衍生物的混合物[16,17],侧链的不饱和度为0~3,其独特的化学结构使漆酚可以发生还原[18]、取代[19]、自聚[20]、共聚[21]等反应。目前生漆主要应用于天然涂料,还未见到有关将漆酚作为色谱分离材料的报道。
在本工作中,以漆酚为原料制备了甲基丙烯酸漆酚酯混合物(UMA),并以UMA为色谱配体,3-(甲基丙烯酰氧)丙基三甲氧基硅烷为硅烷偶联剂,球形硅胶为基质,通过自由基聚合制备了漆酚酯键合硅胶固定相。制备出的固定相不仅具有长烷基侧链,还具有苯环和酚羟基多个活性位点,对天麻和吴茱萸的主要活性成分表现出良好的分离性能。
1 实验部分
1.1 仪器与试剂
Nicolet iS10傅里叶变换红外光谱仪(Nicolet公司,美国); Vario EL cube型元素分析仪(EA, Elementar公司,德国); Z0050716型装柱机(Scientific Systems公司,美国);不锈钢色谱柱(250 mm×4.6 mm,大连依利特分析仪器有限公司); STA449F3热重分析仪(耐驰公司,德国); LC-15C分析型高效液相色谱仪(岛津公司,日本)。
生漆(湖北利川毛坝);球型硅胶(5 μm,苏州纳微科技公司);溴化钾(光谱纯,麦克林试剂公司); 3-(异丁烯酰氧)丙基三甲氧基硅烷(阿拉丁试剂公司),乙腈、甲醇(色谱纯,赛默飞试剂公司);偶氮二异丁腈(AIBN)、十二烷基磺酸钠(SDS)、甲基丙烯酰氯、二甲基丙烯酸乙二醇酯(EGDMA)(阿拉丁试剂公司);天麻(湖北恩施),吴茱萸(广西南宁),天麻素、吴茱萸碱、吴茱萸次碱对照品(分析标准品,≥98%,阿拉丁试剂公司)。三氯甲烷(TCM)、石油醚(PET,沸点60~90 ℃)、无水硫酸钠、三乙胺(TEA)、甲苯等试剂均为国产分析纯。
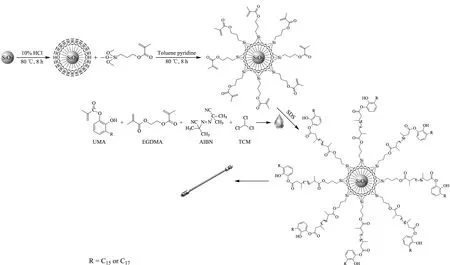
图 2 漆酚酯键合硅胶固定相的合成路线Fig. 2 Schematic illustration of the urushiol methacrylate-bonded silica stationary phase (USP) EGDMA: ethyleneglycol dimethacrylate; AIBN: azodiisobutyronitrile; TCM: trichloromethane.
1.2 漆酚的提取
称取20 g生漆溶于200 mL丙酮中,搅拌使其溶解。将混合液离心后取上层离心液,用丙酮将下层滤渣洗涤至白色,合并离心液。旋蒸浓缩,得到黄棕色油状漆酚混合物12.85 g。
1.3 漆酚酯的制备
漆酚酯的制备过程如图1。称取5 g漆酚混合物于双口烧瓶中,加入100 mL石油醚搅拌使其溶解。称取3.40 g三乙胺和3.33 g甲基丙烯酰氯,分别置于恒压分液漏斗中,在冰水浴条件下滴加到反应体系中。滴加完毕后,升温至40 ℃,在N2保护下回流反应4 h,抽滤,收集滤液并用饱和食盐水洗至中性,旋蒸浓缩后得到黄色油状漆酚酯混合物。
1.4 硅烷化硅胶的制备
称取10 g硅胶(5 μm)于单口烧瓶中,加入100 mL体积分数为10%的盐酸溶液,搅拌均匀。升温至70 ℃,在N2保护下回流反应8 h,减压抽滤并洗涤至中性后在80 ℃条件下真空干燥12 h制得活化硅胶;将活化硅胶置于单口烧瓶中,加入100 mL无水甲苯、4 mL无水吡啶及10 mL 3-(异丁烯酰氧)丙基三甲氧基硅烷,搅拌均匀,升温至80 ℃,在N2保护下回流反应8 h,冷却至室温,减压抽滤并依次用丙酮、甲醇、正己烷洗涤至无吡啶味,最后真空干燥12 h,制备得到硅烷化硅胶。
1.5 漆酚酯键合硅胶固定相的制备
固定相的制备过程如图2。分别称取UMA 0.16 g、EGDMA 0.33 g、AIBN 3.00 mg和0.06 g异
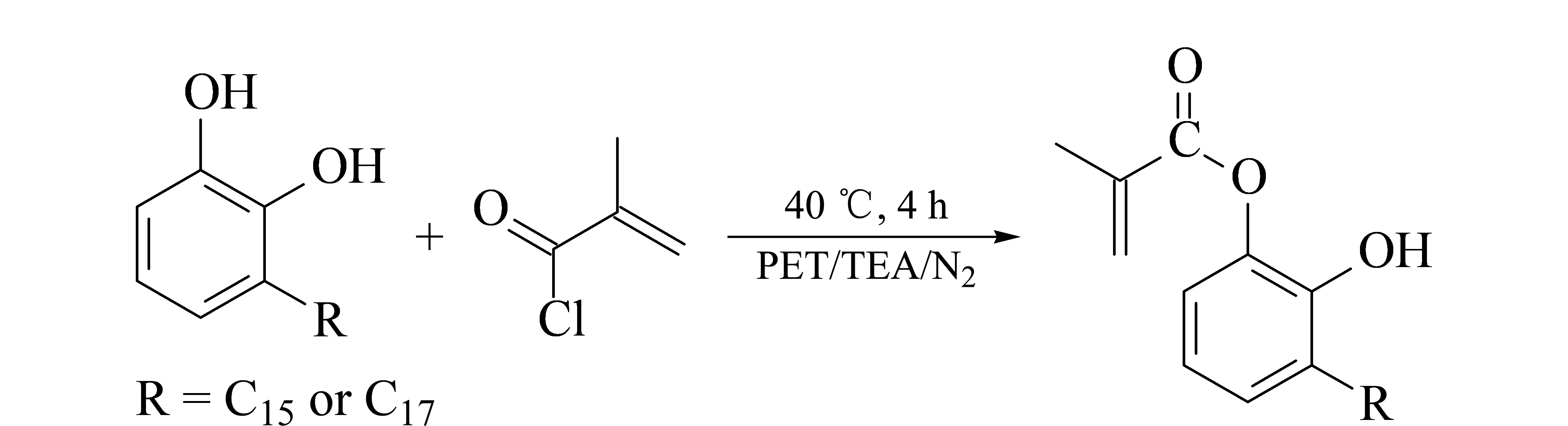
图 1 甲基丙烯酸漆酚酯合成示意图Fig. 1 Synthesis of urushiol methacrylate (UMA)PET: petroleum ether; TEA: triethylamine.
辛烷,合并使其溶解于30 mL的三氯甲烷中得到混合溶液。将混合液超声5 min使其分散均匀。然后将混合液均匀地涂覆到硅烷化硅胶表面。将涂覆后的硅胶均匀分散于100 mL质量浓度为30 g/L的SDS水溶液中进行悬浮聚合。在85 ℃下回流反应2 h,在95 ℃回流反应3 h。反应结束后冷却至室温,抽滤并用无水乙醇进行索氏提取,得到漆酚酯键合硅胶固定相。
1.6 色谱柱的制备
在装柱前,先将制备好的固定相过500目筛。采用匀浆法装柱,以甲醇作为匀浆液,将4 g固定相加入58 mL甲醇中形成匀浆液。以无水乙醇为顶替液,在高压下将固定相填充于不锈钢色谱空柱(250 mm×4.6 mm)中。在60 MPa的压力下装柱15 min,在30 MPa的压力下装柱15 min。装柱完成后,以甲醇为流动相,以0.1 mL/min的流速冲洗色谱柱至基线平稳,即制得漆酚酯键合硅胶色谱柱(USP柱),以甲醇为保护液保存备用。
2 结果与讨论
2.1 甲基丙烯酸漆酚酯的红外光谱
将漆酚与UMA用无水硫酸钠脱水后,均匀涂在KBr镜片上进行红外光谱测试,结果如图3,在3 480 cm-1处的吸收峰为酚羟基峰。酯化产物的酚羟基峰明显减弱,说明酚羟基发生了反应,但由于侧链的空间位阻较大未能将其全部封闭。在1 745 cm-1处有明显的C=O双键的伸缩振动峰,在1 123 cm-1处有明显的C-O-C伸缩振动峰,说明酚羟基与甲基丙烯酰氯发生了取代反应,生成了漆酚酯。
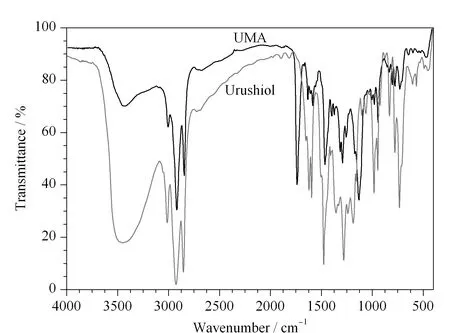
图 3 UMA与漆酚的红外光谱图Fig. 3 Fourier transform-infrared (FT-IR) spectra of UMA and urushiol
2.2 漆酚酯键合硅胶的固定相表征
2.2.1元素分析
分别对硅胶、硅烷化硅胶和漆酚酯键合固定相中的C、H、O元素含量进行测定(见表1)。结果表明,硅烷化硅胶中C、H和O元素的含量分别增加至6.21%、1.25%和6.44%,说明成功制备出硅烷化硅胶。漆酚酯键合固定相的元素分析结果显示,由于漆酚酯中含有C、H、O元素,使得其3种元素含量均有所增加,分别达到8.32%、1.63%和7.73%,证实了漆酚酯通过自由基聚合反应成功聚合在硅烷化硅胶上,表明成功制得漆酚酯键合硅胶固定相。
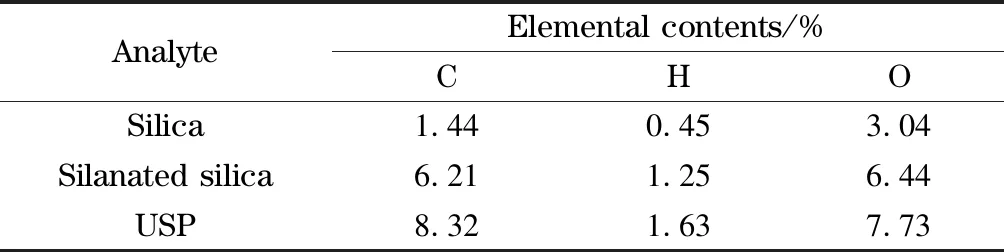
表 1 硅胶、硅烷化硅胶和USP的元素分析结果
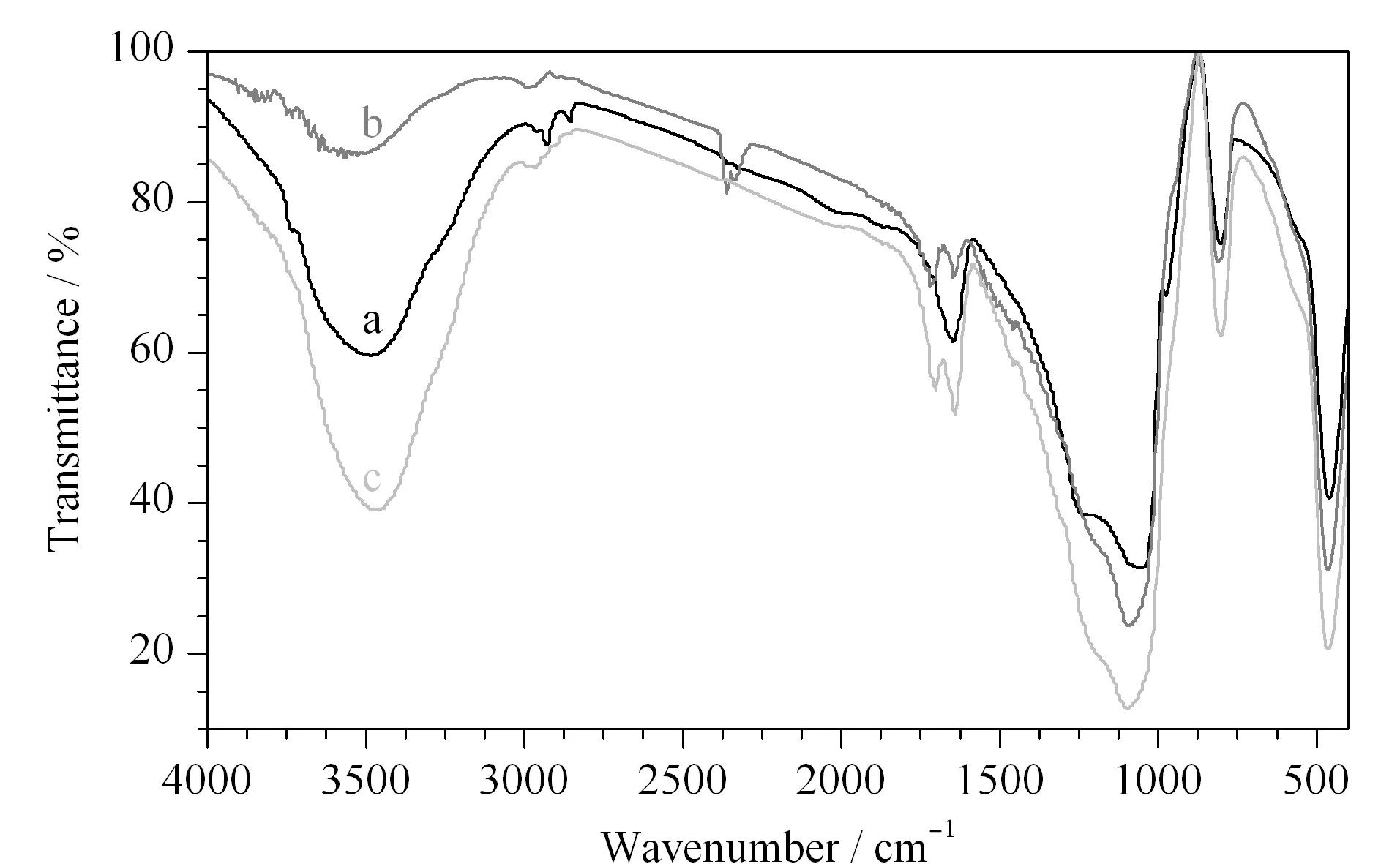
图 4 (a)硅胶、(b)硅烷化硅胶和(c)USP的红外光谱图Fig. 4 FT-IR spectra of (a) silica, (b) silanated silica, and (c) USP
2.2.2红外光谱
硅烷化硅胶与漆酚酯键合硅胶固定相的红外光谱见图4。由图4a可以明显观察到硅胶在3 490 cm-1处有明显的吸收峰,该峰为Si-OH的伸缩振动峰,1 648 cm-1处的吸收峰为硅胶上结合水的H-O-H的弯曲振动峰,1 089 cm-1处的吸收峰为Si-O-Si的反对称伸缩振动峰,972 cm-1处的吸收峰为Si-OH的弯曲振动峰,807 cm-1和458 cm-1处的吸收峰属于Si-O的对称伸缩振动。由图4b可以观察到:与硅胶相比,硅烷化硅胶在3 490 cm-1处的Si-OH伸缩振动峰明显减弱,在972 cm-1处的Si-OH弯曲振动峰明显消失,并在1 704 cm-1处和1 640 cm-1处出现了新的C=O吸收峰和C=C吸收峰,表明成功制备了硅烷化硅胶。由图4c可以明显观察到键合硅胶固定相在3 490 cm-1处、1 704 cm-1处和1 640 cm-1处的吸收峰明显增强,表明漆酚酯成功键合到硅烷化硅胶上。
2.2.3热重分析
通过热重分析评价漆酚酯键合硅胶固定相的键合量和热稳定性。分别称取4 mg左右的待测物质置于Al2O3坩埚内,温度从35 ℃升至800 ℃且升温速率为10 ℃/min,以N2作为保护气,得到热失重曲线(见图5)。可观察到:硅胶(图5a)、硅烷化硅胶(图5b)和键合固定相(图5c)在35~150 ℃出现较小的失重现象,失重率约为1%,这是由待测物质脱水造成的;随着温度的升高,观察到漆酚酯键合硅胶固定相出现明显的热失重现象,而硅烷化硅胶在300 ℃开始分解,硅胶在600 ℃开始分解,说明该段失重现象是由键合到硅烷化硅胶的漆酚酯热分解造成的。在200~800 ℃范围内硅烷化硅胶和键合固定相的失重率分别为7%和12%,表明成功将漆酚酯键合到硅胶上,也证明了该固定相热稳定性良好。
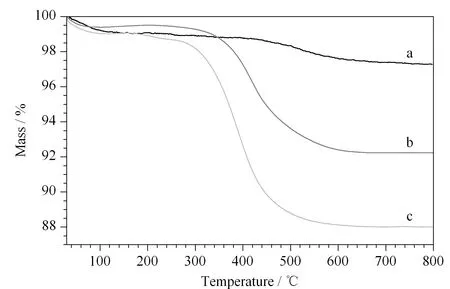
图 5 (a)硅胶、(b)硅烷化硅胶和(c)USP的热失重曲线图Fig. 5 Thermogravimetric (TG) curves of (a) silica, (b) silanated silica, and (c) USP
2.2.4固定相形貌表征
键合固定相的形貌表征如图6。由图6a和图6b可以观察到硅胶与漆酚酯键合硅胶固定相均具有良好的球形结构,粒径在5 μm左右,分布均一,单分散性良好。与硅胶相比,漆酚酯键合硅胶固定相的表面变得粗糙,有明显的附着物,说明漆酚酯成功键合到硅胶上。
2.3 色谱柱对中药主要成分的分离
2.3.1对天麻主要成分的分离
以乙腈-0.05%磷酸溶液(3∶97, v/v)为流动相,柱温25 ℃,进样量20 μL,检测波长220 nm,流速为0.4 mL/min,测试USP色谱柱对天麻浸膏的分离性能(天麻浸膏的制备参考《中国药典》[22])。由图7a可以看出,天麻浸膏经USP柱分离后,出现5个色谱峰,其中一峰与天麻素对照品色谱峰保留时间一致。将流速调至1.0 mL/min,其他色谱条件不变,测试商品化C18柱对天麻浸膏的分离性能,结果如图7b所示,经商品化C18柱分离后出现4个色谱峰,其中一峰与天麻素对照品色谱峰保留时间一致。与商品化C18柱相比,USP柱对天麻浸膏的主要成分实现了基线分离,为天麻素的分离纯化提供了新的思路。

图 6 (a)硅胶和(b)USP的扫描电镜图Fig. 6 Scanning electron microscope images of (a) silica and (b) USP
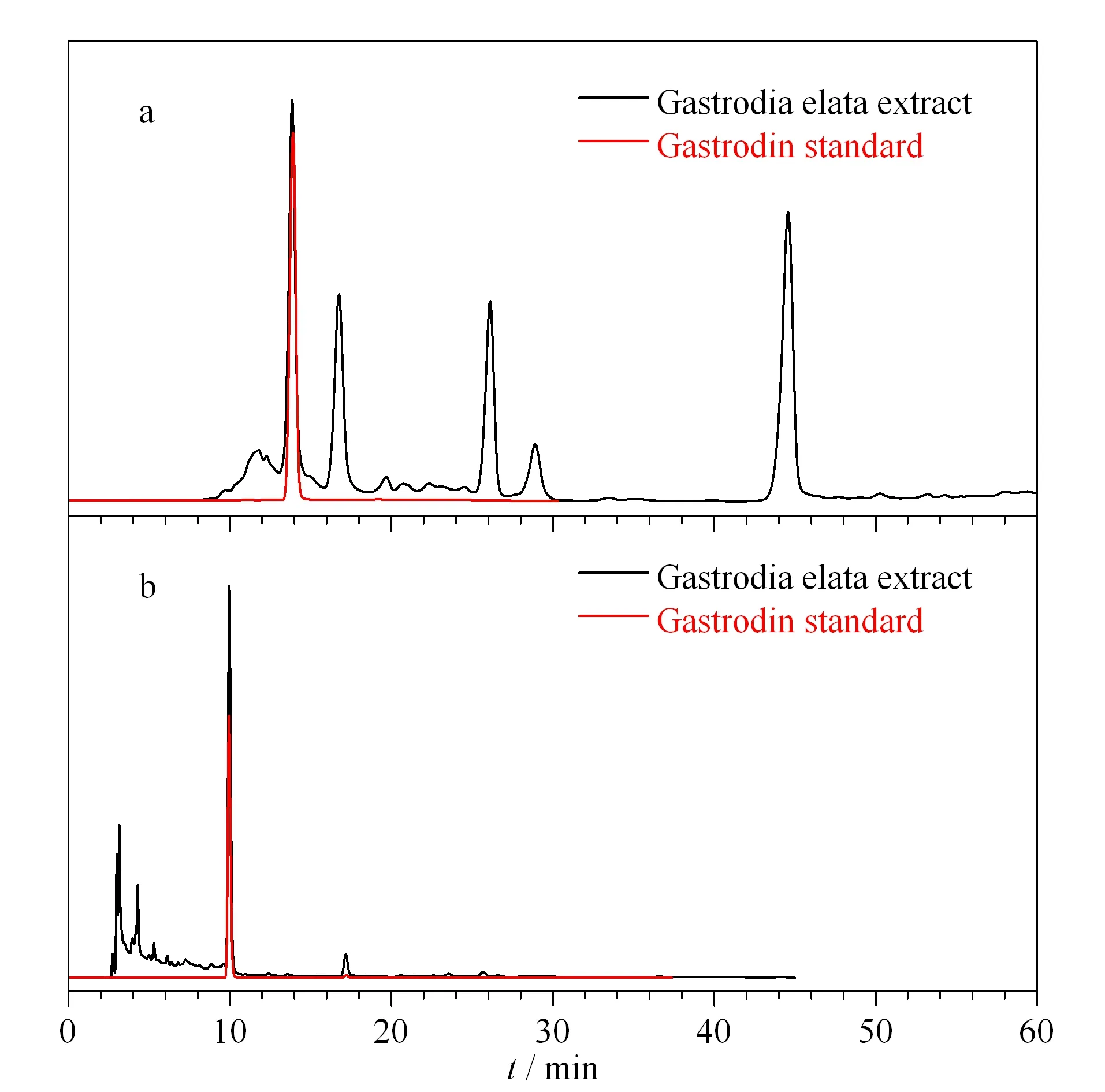
图 7 天麻浸膏在(a)USP柱和(b)C18柱上的色谱分离图Fig. 7 Chromatographic separation of Gastrodia elata extract on (a) USP column and (b) C18 column
2.3.2对吴茱萸主要成分的分离
参考Sun等[15]的方法制备吴茱萸浸膏。
以乙腈-水(50∶50,v/v)为流动相,柱温25 ℃,进样量20 μL,检测波长为290 nm,流速为0.5 mL/min,测试USP色谱柱对吴茱萸浸膏的分离性能。由图8可以看出,浸膏经USP色谱柱分离后,出现2个色谱峰,分别与吴茱萸碱和吴茱萸次碱对照品色谱峰的保留时间一致。而2020版《中国药典》[22]用商品化C18柱分离的流动相为(乙腈-四氢呋喃(25∶15, v/v))-0.02%磷酸溶液(35∶65, v/v)。相对于商品化C18柱,USP柱的分离条件更加安全,简单。
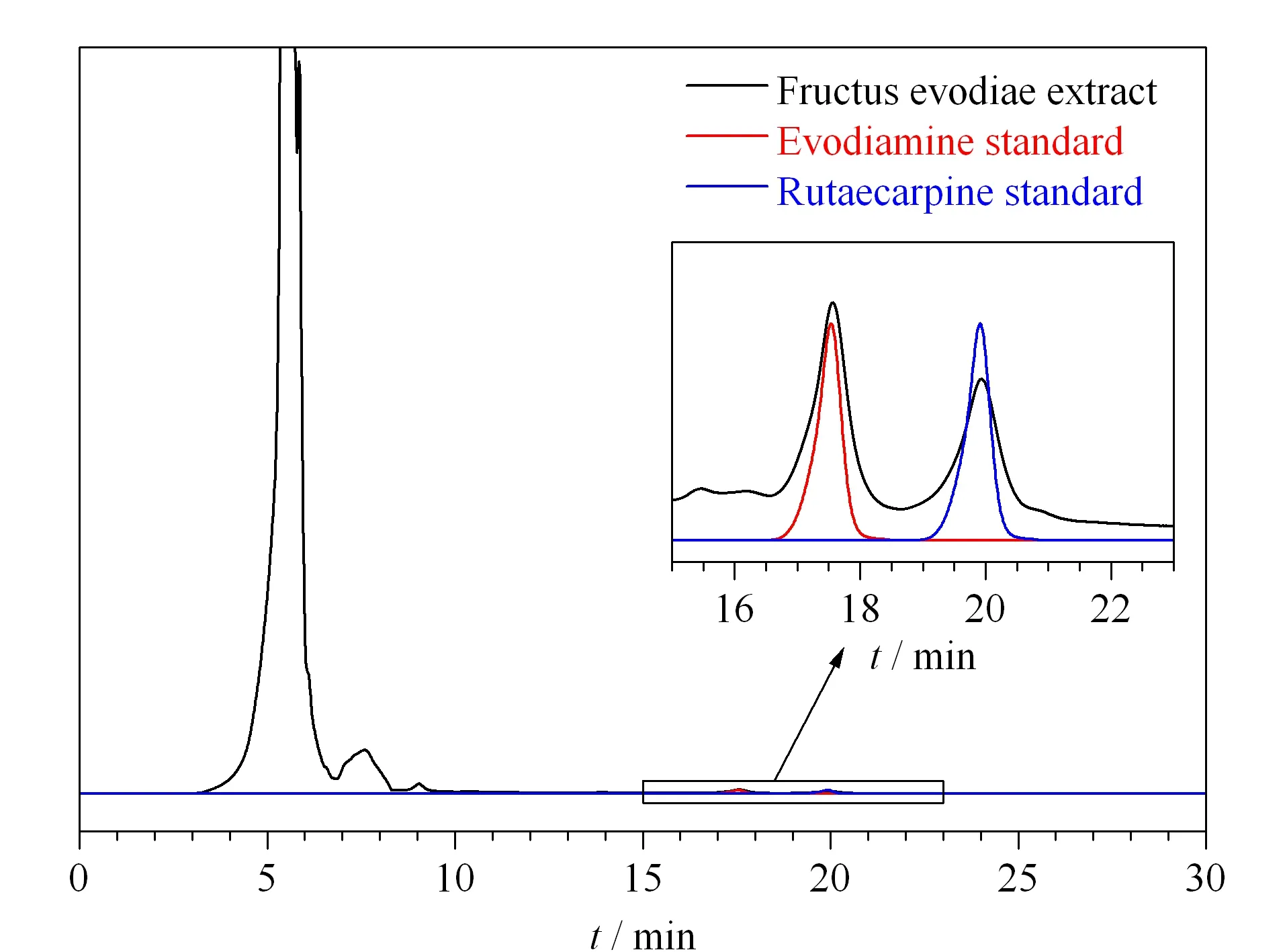
图 8 吴茱萸浸膏在USP柱上的色谱分离图Fig. 8 Chromatographic separation of Fructus evodiae extract on USP column
3 结论
本文以绿色可再生资源漆酚为原料制得UMA,将其均匀涂覆到硅烷化球形硅胶上,通过涂覆-自由基聚合成功制备了一种新型漆酚酯键合硅胶固定相。该固定相对天麻素表现出良好的分离性能,为分离纯化天麻素及天麻的其他活性成分提供了新的途径,有较好的应用前景;同时,该固定相的成功制备为色谱固定相制备提供了新的思路,也扩展了生漆的应用。关于USP色谱柱的性能评价和分离机理研究工作正在进行,后期会将USP填料做成制备柱,并用于天麻素和吴茱萸碱纯化。
