Human embryonic stem cells as an in vitro model for studying developmental origins of type 2 diabetes
2020-09-18
Andy Chun-Hang Chen,Kai Fai Lee,Yin Lau Lee,Department of Obstetrics and Gynaecology,The University of Hong Kong,Hong Kong,China
Andy Chun-Hang Chen,Kai Fai Lee,William Shu Biu Yeung,Yin Lau Lee,Shenzhen Key Laboratory of Fertility Regulation,The University of Hong Kong Shenzhen Hospital,Shenzhen 518053,Guangdong Province,China
Abstract
Key words:Development origins of health and diseases;Maternal diabetes;Environmental insults;Type 2 diabetes;Human embryonic stem cells;Epigenetics
INTRODUCTION
The increasing prevalence of diabetes is a serious global public health concern.According to the latest report from the International Diabetes Federation (Diabetes Atlas 2019),more than 400 million adults are thought to have diabetes[1].More astonishingly,approximately half of them have not been diagnosed.The proportion of people with type 2 diabetes (T2D) has been increasing in most countries,including China.Indeed,the prevalence rate of diabetes in China has increased sharply in recent decades,from 1% in 1980[2]to 9.7% in 2008[3]and further to 10.9% in 2013[4].Another report suggested that only one-fourth of the diabetes patients in China were diagnosed and treated,and among those treated,less than half of them had adequate glycemic control[5].Diabetes is one of the biggest health issues in many countries.There is an urgent need for both national and international entities to tackle this problem.
T2D can be attributed to both genetic and environmental factors.For genetic factors,over 100 loci have been found to be associated with T2D.The susceptibility loci of T2D vary among ethnic groups.For instance,single nucleotide polymorphisms (SNPs) inKCNQ1are associated with T2D in both East Asian and European people[6].ARHGEF11variants increase T2D risks in Pima Indian people[7].On the other hand,SNPs of some loci (TSPAN8-LGR5,THADA,andADAMTS9) are correlated with T2D susceptibility in Caucasian individuals but not in Chinese individuals[8].Association studies suggested that genes such asTCF7L2andKCNQ1were related to pancreatic βcell function and insulin secretion[9,10].However,the causal relationship between genetic variants and disease phenotypes remains largely unclear.For environmental factors,in addition to personal lifestyle,maternal hyperglycemia also contributes to T2D risks.Approximately one-sixth of live births are affected by hyperglycemia during pregnancy[1].Developmental epidemiological[11-13]and animal studies[14,15]indicated thatin uteroexposure to maternal diabetes increased the risks of developing T2D and insulin resistance in offspring.However,mechanistic studies on the inductive action of maternal hyperglycemic conditions on the development of T2D have been confined to animal models or pancreatic cell lines[16,17].With the introduction of human embryonic stem cells (hESCs) in 1999[18],early human embryo development can be studiedin vitro.We and others have used hESCs as models for studying thein uteroeffects of maternal diabetes on early embryo development,which was previously not possible in other pancreatic cell lines.In this review,we will discuss the long-term health consequences of fetal exposure to maternal diabetes and update the use of hESCs for studying the developmental origins of T2D.
MATERNAL CONDITIONS ASSOCIATED WITH DEVELOPMENTAL ORIGINS OF HEALTH AND DISEASES
The concept of developmental origins of health and diseases (DOHaD) was first proposed by Barkeret al[19-21]more than 30 years ago;therefore,it is also known as“Barker’s hypothesis”.The epidemiological studies by Barkeret al[21]revealed a high correlation between infant mortality rate and the incidence of ischemic heart disease later in life.Additionally,fetal malnutrition was associated with the risk of developing heart disease in adulthood[19].Based on their observations,it was suggested that an adverse intrauterine environment would affect fetal programming.These changes permanently shaped the offspring’s organ function and metabolism,which would contribute to the adult onset of noncommunicable diseases (NCDs).Birthweight is the first and most common parameter predicting the health status of individuals at childhood and adulthood.Low birthweight is associated with many NCDs,including heart disease and T2D[19,22].
Maternal malnutrition
Early studies of DOHaD focused on maternal malnutrition.A famous example of this was the Dutch famine study.The offspring cohort who had prenatal exposure to Dutch famine (1944-1945) was traced.Studies have revealed a strong association between prenatal exposure to famine and glucose intolerance[23],obesity[24],heart disease[25],and even breast cancer[26].A follow-up study demonstrated a transgenerational effect leading to neonatal adiposity in the F2 generation from the famine offspring cohort[27].On the other hand,high birthweight,which has become more prevalent recently due to maternal obesity and overnutrition,is correlated with obesity[28],T1D[29],breast cancer,and pancreatic cancer[30].
Maternal exposure to endocrine disrupting chemicals
In uteroexposure to chemicals was found to be detrimental to long-term health in offspring.Animal studies have demonstrated thatin uteroexposure to endocrine disrupting chemicals (EDCs),such as bisphenol A (BPA),alters the development of the mammary gland,increasing the risk of breast cancer[31].Prenatal exposure to BPA and diethylstilbestrol reduces the fertility of female mice,and the effect is transgenerational through the F3 generation[32].In addition to affecting the reproductive system,in uteroexposure to chemicals also contributes to an increase in T2D risk.Prenatal exposure to BPA induced leptin levels in female infants,and elevated leptin levels are correlated with insulin resistance[33].A similar finding was observed in mice,where the administration of low-dose BPA (10 μg/kg) led to the development of chronic hyperinsulinemia and impaired glucose tolerance[34].Another study traced the offspring born from individuals exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) due to explosion incidence in Italy in the 1970s.They found thatin uteroexposure to TCDD increased the risk for metabolic syndrome in male offspring[35].To date,many maternal conditions have been identified to be associated with DOHaD,including maternal stress,hypertension,obesity,diabetes,smoking,infection,malnutrition,and even overnutrition[36].
Maternal diabetes
One-sixth of live births worldwide are affected by hyperglycemia during pregnancy,among which approximately 80% are related to gestational diabetes (GDM)[1].It is therefore apparent that maternal obesity,T2D,and GDM have long-term impacts on offspring health.GDM is defined as women without previously diagnosed diabetes who exhibit high blood glucose levels during pregnancy,especially during the third trimester.The prevalence of GDM ranges from 7%-10% of all pregnancies[37,38].There are several risk factors contributing to GDM,which include obesity and personal or family history of T2D or GDM.Severely obese women have an 8-fold higher risk of developing GDM than pregnant women with a healthy weight[39].It should be noted that GDM not only increased the risks of insulin resistance and T2D in offspring but also in mothers[40].With the increasing number of pregnancies complicated by diabetes,it is important to understand the long-term impacts on offspring health through epidemiological studies.We will discuss the possible mechanisms in the context of epigenetics.
Epidemiological and animal studies:Maternal diabetes is often characterized by increased glucose transport from the placenta to the developing fetus;therefore,fetal macrosomia is the most obvious outcome that is studied[40,41].Macrosomia is defined as birthweight of infants above 90thpercentile of relative gestational age.More than 40%of infants born from diabetic pregnancy develop macrosomia[42],which is associated with increased neonatal morbidity rates.Macrosomic infants have an approximately 5-fold higher risk of glucose infusion and a 2-fold higher risk of neonatal jaundice than healthy infants[43].A similar observation was found in an animal model in which rat offspring born from streptozotocin (STZ)-induced diabetic mothers developed macrosomia[44].The mechanisms by whichin uterohyperglycemia leads to macrosomia are not completely known.It has been suggested that GDM causes downregulation of adiponectin and upregulation of leptin.Macrosomic development has been linked to the modulation of cytokines[45].
The pathologies of macrosomia and maternal diabetes are associated with metabolic defects in infants.Macrosomic infants,and those born from diabetic pregnancies,have altered lipid metabolism.Compared with healthy babies,macrosomic infants have elevated plasma cholesterol and triglyceride levels[46].In an STZ-induced diabetic rat model,the resulting offspring have increased lipid contents in serum and the liver[47].These findings suggest alteration of lipid metabolism in the fetus,which contributes to risks of obesity and T2D in adulthood.Another important metabolic defect in the fetus is insulin secretion.Fetal development in the diabetic environment is accompanied by increased insulin secretion.Hyperinsulinemia has been found in cord blood in mothers with T2D or GDM[48].Increased insulin secretion leads to overstimulation and exhaustion of fetal pancreatic β-cells.There is evidence of degranulation of fetal insulin-producing β-cells in the hyperglycemic intrauterine environment[49].
In addition to metabolic defects,abnormal organ development frequently occurs in offspring exposed to an intrauterine diabetic environment.At the beginning of gestation,impaired gene expression resulting from oxidative stress in the hyperglycemic environment can lead to embryopathy and an increased risk of cardiac,renal,and gastrointestinal malformations[50,51].Early fetal exposure to a diabetic environment is correlated with higher risks of congenital abnormalities than what is observed when analyzing other exposure periods[52].Reduced organ mass is another abnormality observed during development in hyperglycemicin uteroenvironments.In rats born to diabetic mothers,there is a reduction inIgf2expression levels in pancreatic β-cells and a decreased β-cell mass in the fetus[53].In addition,in uteroexposure to hyperglycemia is associated with a reduction in the number of nephrons and alterations ofIgfexpression in the fetal kidney[54,55].
Epigenetic mechanisms:It has long been suggested that epigenetic changes act as mediators between the early life exposure to environmental insults and the later onset of diseases.Epigenetic changes,such as DNA methylation and histone modifications,are actively involved in the course of embryo development.For example,there is global demethylation after fertilization,and DNA methylation is reestablished upon lineage specification[56].Therefore,the early fetal development period is highly susceptible to epigenomic dysregulation with long-term implications for the health of the offspring[57].
The relationship between dysregulation of the DNA methylome and the risk of T2D has been extensively studied.In rats,offspring born from intrauterine growth retardation have increased risks of T2D in adulthood.In these offspring,Pdx1transcription in pancreatic β cells is silenced due to DNA hypermethylation[58].In humans,thePDX1promoter is hypermethylated in the islets of T2D patients and is associated with loweredPDX1expression in islet cells[59].Pdx1is important for early pancreatic specification in mouse embryos[60].Peroxisome proliferator activated receptor gamma coactivator-1 alpha (PPARGC1A),which regulates ATP production,is also hypermethylated in human islet cells from T2D patients,and knockdown ofPPARGC1Adecreased insulin secretion[61].
Two independent studies utilized DNA methylation profiling on islet cells from T2D patients to determine the global dysregulation of the DNA methylome in diabetic pathology.Volkmaret al[62]and Dayehet al[63]reported 254 and 853 differentially methylated genes,respectively,between T2D and normal samples,among which most were hypomethylated in T2D patients.Their studies also indicated that the differentially methylated genes were related to β-cell function,insulin secretion,and T2D pathogenesis.Another report also showed that GDM altered the placental DNA methylome of genes related to insulin signaling and endocrine disorders in both humans and rats[64].
Dysregulation of chromatin modifications is also closely associated with diabetes.High glucose conditions induce p300 acetyltransferase in primary human endothelial cells.The elevated p300 level increases histone acetylation,which results in induced gene expression of vasoactive factors and extracellular matrix proteins such as endothelin-1 (ET-1),vascular endothelial growth factor (VEGF),and fibronectin,leading to functional alterations in endothelial cells mimicking diabetic conditions[65].Histone methylation of the H3K4 active mark and H3K9 repressive mark is responsible for gene expression regulation.In rats,offspring born under diabetic conditions exhibit dysregulated histone modification of thePdx1promoter;there is a progressive loss of H3K4 methylation but a gain of H3K9 methylation on thePdx1promoter,leading to silencing of this gene during development[58].
PLURIPOTENT STEM CELLS AS MODELS FOR STUDYING DOHaD
hESCs have pluripotent characteristics.They can spontaneously differentiate into three germ layers (mesoderm,endoderm,and ectoderm) during embryoid body (EB)formation[18].Directed differentiation protocols of hESCs into specific cell types have been developed.These differentiated cells are excellentin vitromodels for studying early human embryo development.With the introduction of induced pluripotent stem cells (iPSCs) by Yamanakaet al[66]in 2007,advancements were made to the regenerative medicine field,as patient iPSCs could be used to produce specific functional cell types to be used in replacement therapy.Indeed,iPSC technology-based regenerative therapy for diabetes has been vigorously studied in the past 10 years(reviewed in[67]).
Environmental insults such as maternal diabetes have been shown to affect neuronal,cardiac,and pancreatic development in offspring[68,69].There is also evidence indicating the transgenerational epigenetic effects of environmental insults through germ cells.The specific cell lineages differentiated from pluripotent stem cells not only are of benefit for therapeutic purposes but also provide excellentin vitromodels for studying DOHaD and the underlying mechanisms.In this section,we will update the differentiation protocols of those related cell lineages from pluripotent stem cells.The use of the models,in particular the pancreatic cell lineage,for studying the mechanism of DOHaD will also be discussed.
Pancreatic cell lineage
Pancreatic differentiation from hESCs:Since hESCs were first established from human embryos in 1998[18],there have been many studies on the production of glucoseresponsive pancreatic β cells from hESCs for therapeutic purposes.Thein vitroderivation of pancreatic β cells from hESCs involves stepwise inductions of cells representing mesendoderm (ME),definitive endoderm (DE),primitive gut tube (PGT),pancreatic progenitor (PP),and insulin-producing cell (IPC).
The stepwise differentiation of ESCs along the pancreatic lineage requires the activation of different signaling pathways (Figure1).ME cells are bipotent in nature and are able to give rise to both the mesoderm and endoderm lineages during development[70].In an early study of mouse embryonic development,ME cells were found to emerge from the anterior end of the primitive streak (APS)[71].Brachyury (T)[72],goosecoid (GSC)[73],eomesodermin (EOMES)[74],andMIXL1[75]are valuable mesendoderm markers.Activation of the Wnt and TGFβ pathways is important for the derivation of ME cells from hESCsin vitro[72,76].Therefore,the differentiation of ME includes the use of recombinant activin A (AA),which mimics the action of Nodal as the ligand for the TGFβ signaling pathway[77].In addition,treatment with recombinant Wnt3a or a glycogen synthase kinase 3β inhibitor (CHIR-99021) can be used to activate the Wnt pathway[76,78].
DE can give rise to different endodermal cells,such as hepatocytes,epithelial cells of the respiratory tract,and the pancreas[79].The efficient formation of DE cells is essential for subsequent differentiation into functional pancreatic cells[80].The formation of the DE is marked by the expression of several transcription factors,including SRY (sex determining region Y)-box 17 (SOX17)[81],forkhead box A2 (FOXA2),and chemokine(C-X-C Motif) receptor 4 (CXCR4)[82].Similar to ME formation,activation of the TGFβ pathway is important for the induction of DE markers.Recombinant AA and noggin,which acts as a bone morphogenic protein (BMP) antagonist,are used for DE induction[83].Small molecules,including induction of definitive endoderm 1/2(IDE1/IDE2),can mimic the effects of AA.Treatment of hESCs and mESCs with IDE1/2 induces DE formation,which is accompanied by an increase inSOX17expression[84].Using a commercially available DE differentiation kit (STEMdiff DE kit),we have shown that ME cells can be induced after 2 d of differentiation withTandMIXL1,and we have shown the efficient generation of DE cells withSOX17,FOXA2,andCXCR4expression after 5 d of differentiation[85].
The formation of a PGT follows after DE induction[86].Growth factors,including FGF10 and keratinocyte growth factor,enhance the efficiency of PGT formation[76,87].Inhibiting the sonic hedgehog (Shh) signaling pathway by cyclopamine-KAAD treatment efficiently induces PGT specification[76].The action is concordant with inhibition of cells entering an intestinal differentiation pathway following knockout of Shh signaling during mouse pancreatic bud formation[88].Further specification into PP cells requires the continuous activation of FGF and inhibition of Shh signaling.The addition of retinoic acid together with FGF10 and cyclopamine-KAAD enhances PP formation[76].In addition,activation of the protein kinase C (PKC) signaling pathway aids the formation of PP cells from the DE stage.A small molecule,indolactam V,which activates PKC signaling,was found to induce PP differentiation from hESCs[89].The PP cells expressed several markers,includingPDX1,SOX9,NKX6.1,andNKX6.2[76,90].

Figure1 Stepwise in vitro differentiation of pancreatic cells from human embryonic stem cells.Stage-specific markers,regulating pathways,recombinant proteins,and small molecules added at different stages are listed.+:Positive regulation;-:Negative regulation;IDE:Induction of definitive endoderm;Shh:Sonic hedgehog;KGF:Keratinocyte growth factor;RA:Retinoic acid;PKC:Protein kinase C;ILV:Indolactum V;HGF:Hepatocyte growth factor;RSV:Resveratrol.
For the final step of producing IPCs from hESCs,there are two major approaches.One approach is to transplant PP cells into the mouse kidney capsule and allow them to maturein vivo[91].The other approach is thein vitrodifferentiation of IPCs from PP cells.Treatment with extendin 4,hepatocyte growth factor,BMP4,and nicotinamide increased insulin secretion by PP cells in response to high glucose levels.However,thein vitrodifferentiation protocols are not efficient,and only approximately 10% of cells are insulin positive[76,92].Pagliucaet al[93]reported the use of Alk5 receptor inhibitor II,PKC signaling activator,and thyroid hormone in the formation of β cells.Their results demonstrated that the β cells that formed were functional,as transplantation into diabetic mice successfully restored blood glucose to normal levels[93].
Three-dimensional organoid culture methods have recently been developed for the differentiation of hESCs.The organoids formed were reported to be structurally and functionally similar to their native tissue counterparts.For instance,pancreatic organoids were formed by aggregating hESC-derived PP cells in a novel hydrogel system named Amikagel.The resulting cells in the organoids closely mimicked pancreatic islet cells[94].
Pancreatic differentiation from hESCs as a model for studying DOHaD:Diabetic pregnancy is known to increase the risks of insulin resistance and T2D in offspring in adulthood.Epigenetic dysregulation is associated with disease phenotypes.For instance,mice born from diabetic pregnancies exhibit hypermethylation ofpdx1promoter DNA[58].Diabetic pregnancies induce global changes in the DNA methylome related to insulin signaling in the human placenta[64].However,studies on the effects of environmental insults on human fetal pancreas development are very limited.We used hESCs as anin vitromodel to study the developmental origins of diabetes.Early pancreatic differentiation is mainly modulated by histone methylation[95,96].We confirmed that the promoters of DE markers (SOX17,FOXA2,andCXCR4) were marked bivalently by both the activating mark H3K4me3 and the repressive mark H3K27me3 at the pluripotent stage.Upon differentiation into DE,the repressive mark H3K27me3 was removed,leading to active expression of DE markers.More importantly,our study was the first to discover that a hyperglycemic environment disrupted histone methylation patterns,resulting in retention of repressive H3K27me3 marks at DE promoters and a significant reduction in their expression compared to the control.The inhibition of DE specification is also observed in mice uponin uteroexposure to hyperglycemia[85](Table1).Recently,studies have demonstrated active DNA methylation and hydroxymethylation during different stages ofin vitropancreatic differentiation from hESCs.DNA hydroxymethylation has been associated with chromatin accessibility,therefore allowing the binding of transcription factors for efficient pancreatic differentiation[97].The above studies suggest the important roles of DNA methylation and hydroxymethylation in pancreatic development.
In addition to maternal diabetes,the effects ofin uteroexposure to chemicals such as EDCs have also been extensively studied in the later development of offspring.For instance,an animal study showed thatin uteroexposure to BPA increased glucagon secretion in fetal islets by affecting the α-to-β cell ratio[98].Recently,we conductedtranscriptomic and methylomic analyses on hESCs upon low-dose (10 pM) TCDD treatment.Our results revealed that the expression and DNA methylation status of a number of genes were dysregulated upon TCDD treatments.Among them,some of the genes,such as adenosine A1 receptor (ADORA1),ADORA2A,inhibin beta A subunit,and hemopexin,were associated with the pathogenesis of diabetes[99].Lowdose TCDD (10-100 pM) treatment of hESCs also induced hypermethylation of a number of genes that are related to insulin signaling and T2D.Among them,PRKAG1remained hypermethylated even upon PP differentiation.PRKAG1knockdown in the pancreatic cell line INS-1E resulted in elevated levels of secreted insulin[100](Table1).In addition,our findings suggested that the dysregulated DNA methylation patterns induced by early chemical exposure might be maintained during early embryonic development.These changes might lead to pathology,such as insulin resistance and diabetes,in offspring.
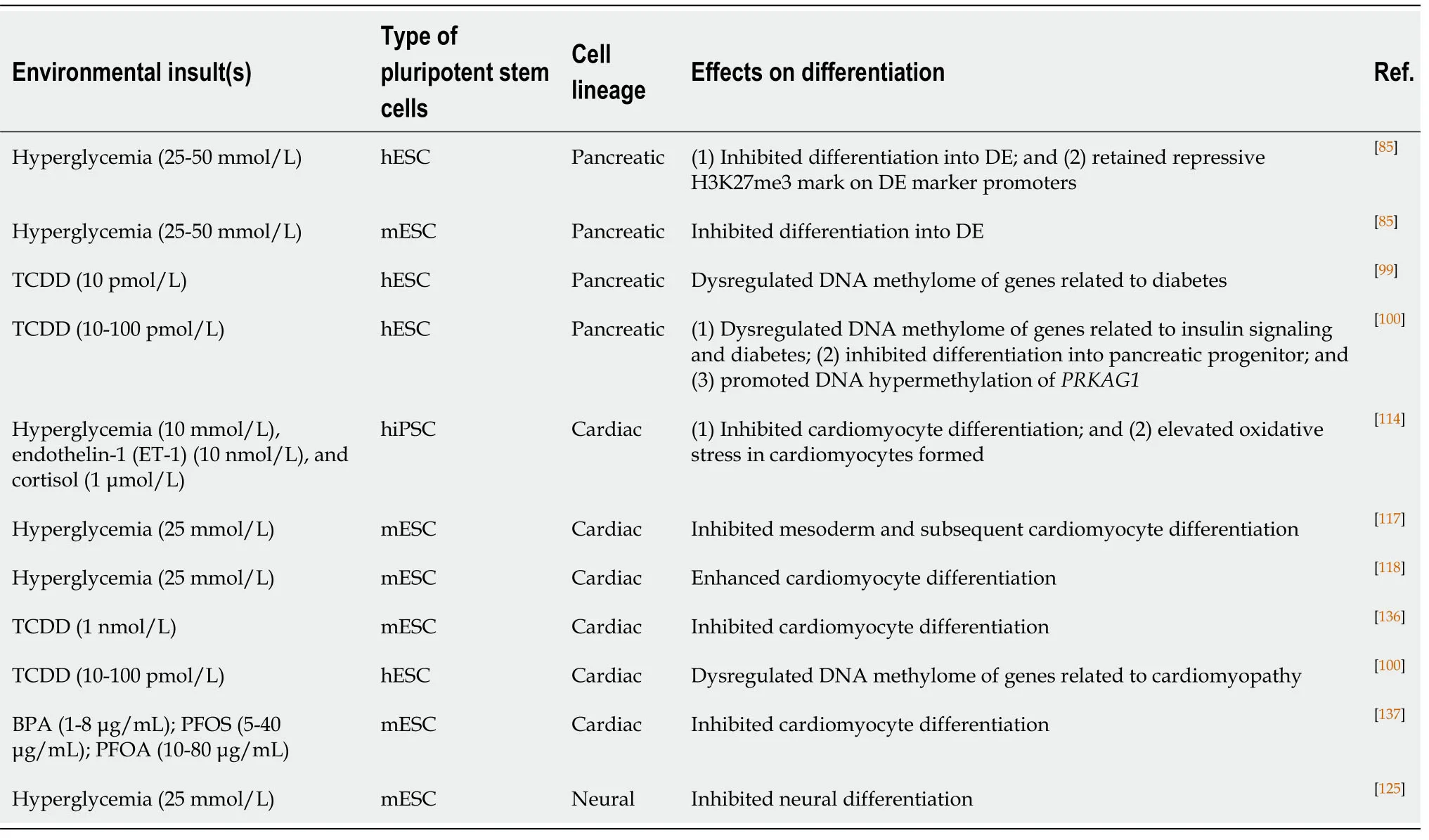
Table1 The use of pluripotent stem cells as in vitro models for studying the developmental origins of health and diseases
Cardiac cell lineage
Cardiac differentiation from hESCs:The human heart is often considered a nonregenerative organ due to the limited proliferative ability of adult cardiomyocytes(CMs).Following the first reports of hESCs[18]and iPSCs[66],several approaches have been developed to differentiate these cells into functional CMs.This section will discuss the transcription factors and cell signaling pathways essential for CM development.We will also introduce various CM differentiation protocols that have been developed.
The heart is one of the organs that develops early in embryos.In the human embryo,the primordial heart begins to develop at 20 d after fertilization.Cardiac cell lineage emerges from the mesoderm.The induction of mesoderm formation is mainly controlled by three cell signaling pathways:the FGF,Wnt and TGFβ pathways.Mesoderm development can be marked with the expression of markers such as T-box transcription factor brachyury (T) andEOMES[101].The mesodermal cell population expressing mesoderm posterior 1 (MESP1) further develop into cardiac progenitor cellsviainhibition of the Wnt/β-catenin pathway[102,103].The subsequent specification into CMs requires the action of signaling pathways such as retinoic acid (RA) and FGF pathways,whereMESP1is the upstream regulator of cardiac-specific transcription factors,such as GATA binding protein 4 (GATA4) and NK2 homeobox 5 (NKX2.5)[104].Thein vitrodifferentiation of hESCs into CMs therefore involves stepwise manipulation of cell signaling pathways.
The successful derivation of CMs from hESCs was first reported through spontaneous differentiation of EBs.However,the efficiency was low,with 8.1% of the area exhibiting spontaneous beating and only 29.4% of cells expressing cardiac troponin I (cTnI) after 20 d of differentiation[105].Several modified protocols have been subsequently reported.These reports also adopted the EB approach,but instead of spontaneous differentiation,and they mimickedin vivosignaling for directed differentiation.For instance,BMP4,Activin A,and bFGF were supplemented in culture for mesoderm induction.VEGF and DKK1 recombinant proteins were then added as Wnt/β-catenin inhibitors.The cells were further treated with VEGF,bFGF,and DKK1 to induce expansion and differentiation into CMs.With improved protocols,the efficiency of CM differentiation was increased (>80%cTnI+vecells),and it was achieved in a shorter period of time (8-10 d)[106,107].Subsequently,different EB culturing tools were developed for scaling up CM production for therapeutic purposes.For instance,microwells allow the production of a large number of uniformly sized EBs[108].On the other hand,researchers developed microcarriers that promoted the expansion of differentiating hESCs in spinner flasks and bioreactors for large-scale CM production[109,110].
Cardiac differentiation from hESCs as a model for studying DOHaD:Diabetic cardiomyopathy (DCM) is a complication of T2D.Maternal diabetes also increases the risk of cardiomyopathy in infants[111,112].An early animal study using streptozotocininduced diabetic mice demonstrated a high rate of apoptosis in cardiomyocytes.Anin vitrostudy using adult CMs also exhibited reduced myofibrillar formation under high glucose treatment[113].However,the underlying mechanisms of the developmental origins of cardiomyopathy remain largely unknown.hESC-derived CMs can therefore serve as an excellentin vitromodel for recapitulating major events during embryonic heart development.
Diabetic conditions,including high glucose (10 mmol/L),ET-1 (10 nmol/L),and cortisol (1 μmol/L) treatments,induce hypertrophic stress with elevated expression of hypertrophic markers (NPPA,NPPB,ACTA1,andMYH7) during CM differentiation from hiPSCs.The treated CMs exhibit cardiomyopathy phenotypes such as disorganized sarcomere structures,accumulation of lipid contents,and oxidative stress[114](Table1).Defects in embryonic CM formation might lead to an increased risk of DCM in adulthood[114].hESC-CMs are not extensively used as a DOHaD model for cardiomyopathy.This could be attributed to the fact that hESC-CMs do not represent fully mature CMs.The contractile function and cardiac marker expression of hESCCMs are not comparable to those of fetal or adult CMs[115,116].Notwithstanding,similar studies have been performed in a mESC model to understand the effects ofin uterohyperglycemia on cardiac development.It was demonstrated that high glucose conditions (25 mmol/L) impaired cardiac differentiation from mESCs compared with what was observed in cells treated with physiological levels of glucose (5 mmol/L).There was a significant reduction in contracting CMs under high glucose levels.In addition,a significant reduction in the expression of mesoderm markers (TandMixl1)and cardiac markers (Gata4andNkx2.5)[117]was observed upon hyperglycemia treatment.However,opposite results from another study showed that CM formation from EBs was more efficient under high glucose treatment[118](Table1).The effects of hyperglycemia and other environmental insults on human CM differentiation require further investigation.
A recent epigenomic study on human CMs revealed that prenatal and postnatal heart development were regulated by DNA methylation and histone modifications.More importantly,active histone marks (H3K27ac,H3K4me3,H3K9ac,and H3K36me3) were found in the promoters of pathology-related genes such as connective tissue growth factor (CTGF) and natriuretic peptides A and B (NPPAandNPPB) in diseased CMs[119].Another recent study demonstrated distinct DNA methylation patterns in atrial and ventricular subtypes of hiPSC-derived CMs[120].These findings reveal that epigenetic regulation not only occurs during prenatal heart development but also is responsible for cardiomyopathy.The study of DOHaD in relation to cardiomyopathy in an epigenetic context warrants further investigation.
Other lineage differentiation from hESCs as a model of DOHaD
Neural lineage:There is a strong clinical association between maternal diabetes and neural tube defects (NTDs).Maternal diabetes increases the risk of central nervous system malformation in fetuses by 10-15-fold over that of nondiabetic mothers[121,122].Similarly,mouse offspring born from diabetic mothers have an approximately 10%chance of developing NTDs.A high level of oxidative stress leads to neural cell apoptosis in the affected offspring[123].Maternal hyperglycemia also results in the activation of apoptosis signal-regulating kinase 1 (Ask1) in the developing neural tubes of mouse embryos.The activation ofAsk1is related to an increase in caspase 8 protein levels and apoptosis[124].
Studies in animal models provide information on the effects of thein uteroenvironment on early neural development.However,further studies remain challenging because of the limited number of cells in fetal neural tissues.Nevertheless,high glucose treatment (25 mmol/L)in vitroimpedes neural differentiation,resulting in the downregulation of neuronal markers (Sox1,Nestin,andPax6)[125](Table1).Folate deficiency was shown to induce inhibition of the DNA methylation cycle,leading to NTDs in animals[126].Knockout of histone modifiers such asSirt1and histone deacetylase 4 also causes NTDs in developing mouse embryos[127,128].It should be noted that the effects of environmental insults on human neural development may be different from those observed in mice.Further mechanistic studies using hESCs as cell models can improve our understanding in the context of DOHaD.Indeed,treatment with noggin,which inhibits the BMP pathway,successfully enabled derivation of neuronal cells from hESCs.The neurospheres formed could further differentiate into mature neurons and glia[129],providing a good cellular research model.
Germ cell lineage:Growing evidence suggests that the negative impacts of adverse intrauterine environments on offspring might be transgenerational,meaning that the disease phenotypes will be expressed in the F2 generation.Such transgenerational effects are evidenced in animal models.For example,vinclozolin (VCZ;3-(3,5-dichlorophenyl)-5-methyl-5-vinyl-oxazolidine-2,4-dione),one of the EDCs widely used as a fungicide,dysregulates the epigenome of primordial germ cells (PGCs) in mice from F1 to F3;the microRNA pattern in F1-F3 PGCs is disrupted following F0 animal exposure to VCZ[130].The downregulation ofmiR-23bandmiR-21in the treated mice disrupts the let7 pathway,leading to increased apoptosis of embryonic PGCs.Another EDC,TCDD,alters transcriptomes in the gonads of F1 and F2 zebrafish[131].The genes with altered expression were related to lipid and glucose metabolism,oxidative stress,and sperm cell development.
In addition to EDC exposure,the transgenerational effects of maternal hyperglycemia have been extensively studied in animal models.Dinget al[132]reported that the mating of F1 male mice from diabetic pregnancies with normal female mice resulted in F2 mice with increased birth weight and impaired glucose tolerance.They associated the above observations with DNA hypermethylation of imprinted genesIgf2andH19in the pancreatic islets of F1 and F2 mice[132].A recent report revealed that maternal diabetes dysregulated the DNA methylome of embryonic F1 PGCs.The differentially methylated genes were related to obesity,insulin resistance,and T2D.More importantly,the same pattern was also observed in F2 somatic cells[133].These studies demonstrated that environmental insults,such as chemicals or hyperglycemia,could be transmitted transgenerationally by changing the epigenomes of germ cells.
In vitrogerm cell differentiation from ESCs has only recently been reported.Haploid germ cells can be generated by coculturing mESC-derived PGC-like cells with neonatal testicular somatic cells.In vitro-derived haploid spermatids are able to generate offspring when injected into oocytes[134].In human culture systems,PGCs can be successfully derived from hESCs.The derivation protocol adopted a stepwise approach recapitulatingin vivodevelopmental events,where Wnt and BMP pathways were activated to drive the formation of premesodermal cells.The specifications of PGCs were then induced by treatment with growth factors such as BMP2,stem cell factor,and epidermal growth factor[135].Advances in germ cell differentiation using a human cell model also provide an opportunity for the study of the transgenerational effects of DOHaD.
CONCLUSION
Much evidence supporting the idea of DOHaD has been obtained from animal models and observational studies of human.The mechanisms behind the long-term health consequences of fetal exposure to adverse maternal conditions are largely unknown in humans.Accumulating data from both hESCs and mESCs suggested that early cell lineage differentiation might be one of the vulnerable embryonic windows through which early exposure to adverse maternal conditions could exert its diabetogenic effects.While protocols for the differentiation of different cell types from ESCs still require further improvement to better mimic physiological development,it is expected that the information obtained from these cell models will provide valuable mechanistic insight into the mechanisms underlying the DOHaD.
杂志排行

World Journal of Stem Cells的其它文章
- Mesenchymal stromal cells as potential immunomodulatory players in severe acute respiratory distress syndrome induced by SARS-CoV-2 infection
- Practical choice for robust and efficient differentiation of human pluripotent stem cells
- Stem cell therapy for Alzheimer's disease
- Exosomes derived from stem cells as an emerging therapeutic strategy for intervertebral disc degeneration
- Off-the-shelf mesenchymal stem cells from human umbilical cord tissue can significantly improve symptoms in COVID-19 patients:An analysis of evidential relations
- Autophagy in fate determination of mesenchymal stem cells and bone remodeling