Early-onset refractory diarrhea due to immune dysregulation,polyendocrinopathy,enteropathy,X-linked syndrome associated with a novel mutation in the FOXP3 gene:A case report
2020-09-14NaSuChengChenXiaZhouGuoDaMaRiLingChenChuanTian
Na Su,Cheng Chen,Xia Zhou,Guo-Da Ma,Ri-Ling Chen,Chuan Tian
Na Su, Department of Pediatrics,Shunde Women and Children's Hospital,Guangdong Medical University,Foshan 528300,Guangdong Province,China
Na Su,Department of Pediatrics,Guangdong Medical University,Zhanjiang 524000,Guangdong Province,China
Cheng Chen,Ri-Ling Chen,Department of Pediatric Internal Medicine,Shunde Women and Children's Hospital,Guangdong Medical University,Foshan 528300,Guangdong Province,China
Xia Zhou, Department of Neurology,Guangdong Medical University,Zhanjiang 524000,Guangdong Province,China
Guo-Da Ma,Maternal and Child Research Institute,Shunde Women and Children's Hospital,Guangdong Medical University,Foshan 528300,Guangdong Province,China
Chuan Tian, Department of Pediatric Hematology,Affiliated Hospital of Guangdong Medical University,Zhanjiang 524000,Guangdong Province,China
Abstract
Key words:Immune dysregulation,polyendocrinopthy,enteropathy,X-linked syndrome;Forkhead box protein 3;Mutation;Refractory diarrhea;Regulatory T cells;Case report
INTRODUCTION
Immune dysregulation,polyendocrinopthy,enteropathy,X-linked(IPEX)syndrome is a rare disorder of genetic autoimmunity caused by mutations in the forkhead box protein 3(FOXP3)gene[1].FOXP3 is an important transcription regulator in the development of CD4+CD25+regulatory T(Treg)cells.FOXP3gene mutations lead to developmental defects,differentiation disorders,and dysfunction of Tregs,which lead to serious autoimmune phenomena in multiple systems[2].The disease mainly influences the intestine,endocrine system,and skin[2],whereas the thyroid glands,kidneys,blood cells,liver,and joints may also be influenced[3].IPEX is an X-linked recessive disorder,and patients who have not been treated effectively in the early stage mostly die within 2 years of age[4].The main treatment measures for patients with IPEX include supportive care,immunosuppressive treatments,and hematopoietic stem cell transplantation.The clinical manifestations of the disease were first reported in 1982 by Powellet al[5],and an increasing number of cases were subsequently reported.Here,we report a male patient with IPEX syndrome who had refractory diarrhea,failure to thrive,hypothyroidism,and nephrotic syndrome and who was hemizygous in theFOXP3gene[c.542G>A(p.Ser181Asn)].
CASE PRESENTATION
Chief complaints
A 13-month-old male patient was admitted to the hospital in June 2019 due to chronic diarrhea and malnutrition.
History of present illness
The patient's symptoms started a year ago with recurrent diarrhea and malnutrition.His diarrhea had been worsened the last 4 h(10 watery bowel movements).
Personal and family history
The patient is the second live-born child of nonconsanguineous and healthy parents(Figure 1)and was born at 35 wk of gestation by spontaneous vaginal delivery.The newborn had a birth weight of 2.15 kg and was hospitalized as a premature infant.The family history was negative for inherent diseases.
History of past illness
At the age of 1 mo,the patient had refractory diarrhea(up to 10-15 watery bowel movements per day)and scattered eczema.Therefore,he was hospitalized on several occasions at a local hospital and given hydrolyzing protein formula milk and supportive care to improve gastrointestinal symptoms.The eczema gradually disappeared in May after birth,but diarrhea did not significantly improve.
Physical examination
Upon admission to the hospital,the patient had a weight of 6.4 kg,head circumference of 40.5 cm,and length 70 cm,failure to thrive,and moderate-severe dehydration.Abdominal subcutaneous fat thickness was less than 4 mm,and bowel sounds were active,approximately 7 per minute.
Laboratory examinations
The laboratory results showed a normal eosinophil count and hemoglobin concentration;a high white blood cell count(23.25×109/L);normal immunoglobulin A,IgG,and IgM levels but markedly elevated IgE levels(1970 IU/mL);high triglyceride(3.74 mmol/L);and decreased complement C3(0.25 g/L),complement C4(0.1 g/L),and albumin(29.1 g/L).The endocrine test showed normal antithyroid peroxidase(aTPO),antithyroglobulin(aTG),and thyroid stimulating hormone(TSH),and high free triiodothyronine(FT3,2.20 pmol/L)and free thyroxine(FT4,10.49 pmol/L)(Table 1).Multiple urinalyses indicated proteinuria(+ to +++).Multiple stool analyses were unremarkable.The patient was allergic to eggs,peanuts,and milk.Endoscopy(gastroscope and colonoscopy)found duodenal anterior wall erosion,and the immunohistochemical staining for cytomegalovirus(CMV)andin situhybridization for EBV-encoded small RNA(EBER)were negative.Flow cytometry showed a slightly higher proportion of CD25+FOXP3+Tregs in CD4+T cells(13.0%)(Figure 2).
Further diagnostic work-up
The patient had early refractory diarrhea,malabsorption,high immunoglobulin E,hypothyroidism,and nephrotic syndrome.The diagnosis of IPEX was confirmed by whole exome sequencing,which revealed a pathogenic site in exon 5,c.542G>A(p.Ser181Asn)mutation of theFOXP3gene.Subsequently,the same heterozygous mutation was found in the mother,who was a carrier,but his father and older brother were normal(Figure 3).The family pedigree of the patient is shown in Figure 1.
FINAL DIAGNOSIS
The final diagnosis of the presented case was IPEX syndrome,hypothyroidism,and nephrotic syndrome.
TREATMENT
The patient was initially diagnosed with refractory diarrhea,malabsorption,hypothyroidism,and nephrotic syndrome.He was treated with anti-infection medications,total parenteral nutrition,prednisone(5 mg/d),and euthyrox(37.5µg/d).After 10 d of this treatment,he had 4-6 watery bowel movements per day but was not significantly gaining weight.Subsequently,the diagnosis of IPEX was confirmed by whole exome sequencing,and a scheduled immunosuppressor treatment was started.However,the family thought that these drugs had severe side effects and refused this type of treatment.Therefore,the patient was treated with prednisone,euthyrox,and other supportive cares.
OUTCOME AND FOLLOW-UP
Since the diagnosis of IPEX syndrome,the patient has been regularly treated with prednisone and euthyrox.Instead of getting better,his condition is slightly worse.He was hospitalized for refractory diarrhea(up to 15-20 watery bowel movements per day)and anasarca,and was discharged after correction of electrolyte disorder and dehydration therapy.At present,he is awaiting hematopoietic stem cell transplantation.

Figure 1 Family pedigree.The black square and arrow indicate the patient,the point inside the circle indicates a heterozygous carrier of the gene,and white squares represent normal members in the family.
DISCUSSION
The typical symptoms of IPEX syndrome present as early refractory diarrhea,type 1 diabetes mellitus,and eczema[2].The enteropathy of IPEX syndrome is present in virtually all affected individuals.The main manifestation of the patients was watery diarrhea and sometimes mucoid or bloody diarrhea[2].Dermatitis is most frequently eczematous[6]and can also be psoriasiform[7]and ichthyosiform[8].Endocrine diseases are common in type 1 diabetes mellitus,most of which are newborn[9],but hypothyroidism has also been reported[3].Rare symptoms such as thrombocytopenia,hepatitis,pneumonia,and nephrotic syndrome have also been reported[3,10].In this report,we present an early-onset IPEX syndrome case with chronic diarrhea as the first symptom at the age of 1 mo,as well as hypothyroidism and nephrotic syndrome.The patient received anti-infection therapy,total parenteral nutrition,and other supportive care more than five times after admission in 1 year.However,diarrhea was not well controlled.Finally,the patient was diagnosed with IPEX syndrome associated with a novelFOXP3gene mutation[c.542G>A(p.Ser181Asn)] by whole exome sequencing.
The humanFOXP3gene is located on the X chromosome(Xq11.23-Xq13.3)and consists of 11 coding exons plus one upstream non-coding exon[11].The gene encodes a protein of 431 amino acids,which contains a repressor domain,a zinc-finger(ZF)domain,a leucine zipper(LZ)domain,and a forkhead(FKH)domain(Figure 4).Ziegleret al[12]found that there were two functional domains within the N-terminal of FOXP3,one between amino acids 67 and 132 and the other between amino acids 135 and 198,which are functional as transcriptional repressors;the latter is especially required for inhibition of nuclear factor of activated T cells(NFAT)-mediated transcription.NFAT is an important transcription factor of cytokine gene expression.In the promoters of cytokine genes,it was found that the NFAT site potentially overlapped or was adjacent to the FKH-binding site[12].Therefore,FOXP3 may play an immunosuppressive function by competing for the DNA-binding sites of NFAT or directly inhibiting the negative feedback of NFAT activity to regulate the expression of cytokine genes[13,14].Therefore,this mutation leads to impaired NFAT transcriptional regulatory activity by altering the binding sites to DNA or inhibiting cytokine gene expression and eventually is responsible for this disease.
TheFOXP3gene is a master regulator of Tregs[2].IfFOXP3mutation leads to this protein being functionally defective,it is unable to sustain Treg development[15].Therefore,most IPEX patients had decreased percentages of CD25+FOXP3+Tregs in CD4+T cells.However,the patient had increased percentages in this study.In previous reports,in addition to the natural Treg cells in the thymus,peripheral CD4+CD25-cells can also develop into Tregs with inhibitory function by stimulation of anti-CD3 and anti-CD28[16]or by retroviral vector or transgene expression.Bacchettaet al[17]found that the ratio of CD4+CD25+Tregs in peripheral blood mononuclear cells(PBMCs)can appear normal,but their capacity to suppress was impaired,which indicates that theFoxp3mutation could not affect Treg development but affected suppression function.Therefore,the function ofFOXP3is not restricted to Tregs but it could also play a role outside the Treg subset.It has been reported in the literature that the percentage of CD4+CD25+FOXP3+Tregs in CD4+T cells or the percentage of FOXP3 expressing CD4+cells in children with IPEX is high(for example,mutation sites were p.M370I[18],p.E323K[19],and p.S390N[19]).Therefore,we speculate that the high ratio of CD25+Tregs in our patient may be related to other regulatory factors.

Table 1 Hematologic and immunologic data of the immune dysregulation,polyendocrinopthy,enteropathy,X-linked patient
CONCLUSION
IPEX is a rare X-lined recessive disease with multiple system involvement,especially enteropathy and refractory diarrhea,caused by mutations in theFOXP3gene.The onset of symptoms is as early as the first few months of life,and patients usually require supportive and alternative treatments.In the differential diagnosis of any patient with unexplained and intractable diarrhea,the disease should be considered and eventually confirmed by gene testing.

Figure 2 Flow cytometry showing CD25+FOXP3+ regulatory cells in the CD4+T cell population in peripheral blood.The red number in each quadrant indicates the percentage of the individual.The percentages of CD25+FOXP3+T cells of the patient and his mother were not reducing(reference range 5.1%-12.7%).
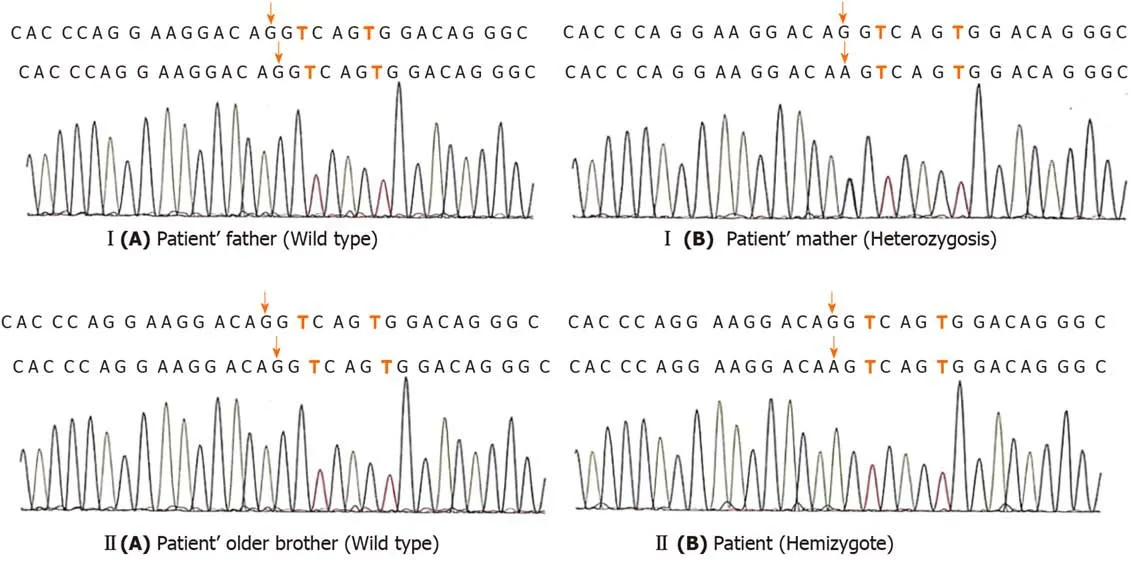
Figure 3 The gene sequencing peaks of the two-generation family.I(A)and II(A)indicate that no mutation was found;I(B)and II(B)indicate that the mutation site is c.542G> A.

Figure 4 Schematic representation of the FOXP3 gene and protein.Numbers along the top line indicate amino acid number(1-431).The latter two lines refer to the gene structure,the coding and non-coding regions:N-terminal domain(orange),zinc-finger(ZF)domain(green),leucine-zipper(LZ)domain(blue),LZ-FKH loop(yellow),and FKH domain(red).The orange arrows indicate the mutation site identified in this study.
杂志排行

World Journal of Clinical Cases的其它文章
- French Spine Surgery Society guidelines for management of spinal surgeries during COVID-19 pandemic
- Prophylactic and therapeutic roles of oleanolic acid and its derivatives in several diseases
- Macrophage regulation of graft-vs-host disease
- Antiphospholipid syndrome and its role in pediatric cerebrovascular diseases:A literature review
- Remotely monitored telerehabilitation for cardiac patients:A review of the current situation
- Keystone design perforator island flap in facial defect reconstruction