基于QuEChERS液相色谱-串联质谱法测定茶叶中除虫脲残留量的不确定度评定
2020-09-11覃慧丽
武 源,覃慧丽
除虫脲又名敌灭灵,是一种苯甲酰胺类杀虫剂,具有对人畜低毒、对环境无公害等特点,其已被广泛用于茶园害虫的防治[1-5]。茶叶产业关乎我国农业农村发展,茶叶质量安全影响我国茶叶对外贸易,近年来,农药残留已成为影响茶叶质量安全的重要因素之一,除虫脲及其降解产物残留问题日益突出[6-8],严重影响到茶叶质量安全。我国国家标准GB 2763—2019《食品安全国家标准食品中农药最大残留限量》规定茶叶中除虫脲的最大残留限量为20 mg/kg,日本和欧盟等进口国家或地区均对茶叶制定了严格的农药残留限量[9-10],日本规定除虫脲在茶叶中最大残留限量为20 mg/kg[11]。由于茶叶样品基质复杂,含有大量的生物碱、色素、咖啡因、茶多酚等干扰物质,使得农残检测难度增大。为确保分析结果的准确性,对检测结果进行不确定度评定是必要的,不确定度评定对于检测过程的质量把控具有重要意义[12]。
目前除虫脲在液相色谱-串联质谱(LC-MS/MS)法的不确定度判定研究基本空缺。本研究依据文献[3-16],对测定茶叶中除虫脲残留量进行了分析与评定,明确了QuEChERS液相色谱-串联质谱法测定茶叶中除虫脲残留量不确定的主要影响因素,为评价除虫脲残留量测定结果的准确性和可靠性提供技术依据。
1 材料与方法
1.1 材料与试剂
提取试剂:4 g MgSO4,1 g NaCl,0.5 g三水合二柠檬酸二钠,1 g柠檬酸钠及两颗陶瓷均质子,美国Agilent公司;净化试剂管:1 200 mg MgSO4,400 mg PSA,400 mg C18,200 mg GCB,美国Agilent公司。
除虫脲对照品(纯度99.65%),德国Dr. Ehrenstorfe公司;三苯基磷酸酯对照品(TPP,纯度99.6%),德国Dr.Ehrenstorfe公司;甲醇、乙腈(均为色谱纯),德国Merck公司;甲酸、乙酸铵(均为色谱纯),德国Fluka公司。
1.2 仪器与设备
Xevo TQ-S 液 质 联 用 仪、ACQUITY UPLCBEH C18色谱柱(2.1×50 mm,1.7 μm),美国WATERS公司;XP26微量电子天平,瑞士Mettler Toledo公司;IKA VORTEX 4 basic涡旋振荡器,德国IKA公司;TTL-DC Ⅱ型氮吹仪,北京同泰联科技发展有限公司;SIGMA 3-3OK离心机,德国SIGMA公司;Milli-Q超纯水设备,美国Millipore公司。
1.3 方法
1.3.1 标准溶液的配制
阴性基质溶液:称取不含目标物的茶叶按前处理步骤处理即得。
除虫脲标准储备液(500 μg/mL):精确称取5 mg除虫脲对照品于10 mL的容量瓶中,用乙腈溶解并定容至刻线。
TPP内标储备液(500 μg/mL):精确称取5 mg内标物对照品TPP于10 mL的容量瓶中,用乙腈溶解并定容至刻线。
除虫脲标准工作液(500 ng/mL):精确吸取0.02 mL的除虫脲标准储备液于20 mL容量瓶中,用乙腈定容至刻线。
TPP内标工作液(5 μg/mL):精确吸取0.2 mL的内标储备液于20 mL容量瓶中,用乙腈溶解定容至刻线。
基质标准工作液:用移液枪分别精确移取0.01、0.02、0.05、0.10、0.20、0.50 mL的除虫脲标准工作液于10 mL的氮吹管中,加入0.01 mL内标工作液,氮吹近干,准确加入1.0 mL阴性基质溶液,涡旋复溶,即得基质标准工作液。以系列标准溶液中除虫脲与内标TPP质量浓度的比值(C除虫脲/CTPP)为横坐标,以除虫脲与内标TPP定量离子峰面积的比值(A除虫脲/ATPP)为纵坐标,绘制基质标准曲线。
1.3.2 提取
茶叶粉碎后进一步充分混匀,准确称取2 g(精确至0.01 g)茶叶置于50 mL具塞离心管中,加10 mL水涡旋混匀,静置30 min。离心管中加入0.10 mL TPP内标工作液,10 mL乙腈,加盖涡旋振荡30 s后加入提取试剂,立即剧烈振荡1 min,防止结块。以转速4 000 r/min离心5 min,待净化。
1.3.3 净化
取8 mL乙腈提取液转移至15 mL净化试剂管中,盖紧离心管,充分涡旋振荡1 min,以转速4 000 r/min 离心5 min,净化液过0.22 μm滤膜后,取1 mL氮吹近干,加入1 mL水-甲醇(9∶1,V/V)涡旋混匀后上机分析。
1.3.4 LC-MS/MS条件1.
3.4.1 液相条件
1.3.4.2 质谱条件
电喷雾离子源(ESI);多反应监测(MRM)采集模式,正离子扫描。目标物和内标的保留时间、定量离子对和定性离子对见表1。

表1 化合物保留时间及离子对
1.4 数学模型的建立
茶叶中目标物残留量X按公式(1)计算:

式中:X为茶叶中除虫脲的残留量,mg/kg;C0为从基质标准工作曲线得到的茶叶溶液中除虫脲的质量浓度,ng/mL;V为茶叶经前处理后的稀释体积,mL;m为称取茶叶质量,g。
2 结果与分析
2.1 称量标准物质及配制基质标准曲线时产生的不确定度μr(2.1)
2.1.1 除虫脲纯度产生的不确定度μr(P)
除虫脲的纯度为99.65%,查阅除虫脲对照品标准证书,其不确定度U95为0.30%,在95%置信水平下,k=2,则标准不确定度μr(P)=0.15%,相对标准不确定度μr(P)=0.001 5。
2.1.2 除虫脲称量产生的不确定度μr(M)
精密称量除虫脲对照品5.757 mg,用乙腈稀释并定容至10 mL,得到除虫脲标准储备液。微量电子分析天平校准不确定度为主要分量,根据分析天平检定证书,本试验所用天平的最大允许误差为0.005 mg,按照均匀分布计算则除虫脲标不确定度μr(M)=0.002 9 mg,相对标准不确定度μr(M)=0.000 5。
2.1.3 配制基质标准曲线时产生的不确定度μr(V)
根据JJG196—2006《常用玻璃量器》[17]检定规程的A级最大允许误差(以下玻璃量器均按A级进行评定)及广西壮族自治区计量检测研究院提供的实验室移液枪的检定证书,按照式(2)计算量器容量允差产生的相对标准不确定度μr(V校准):

式中:a校准为玻璃器具校准引起的体积偏差;k取平均分布,即
实验室环境温度波动范围为(20±5)℃,20℃乙腈的体积膨胀系数为1.37×10-3℃-1,根据式(3)计算室温波动引起的相对标准不确定度μr(V温度):

式中:a温度为实验环境温度波动带来的溶剂体积偏差;k取平均分布,即
分别移取0.01、0.02、0.05、0.10、0.20、0.50 mL的除虫脲标准工作液于10 mL的氮吹管中,加入0.01 mL内标工作液,氮吹近干,用移液枪准确加入1 mL阴性基质溶液,充分涡旋。实验中配制基质标准曲线时移取和定容步骤,用到不同规格的移液枪和容量瓶,均按平均分布计算各分量合成配制基质标准曲线时产生的不确定度μr(V)(表2)。
基质标准曲线稀释过程由上述19次移液,2次定容完成,配制基质标准曲线时产生的不确定度μr(V)=0.021。
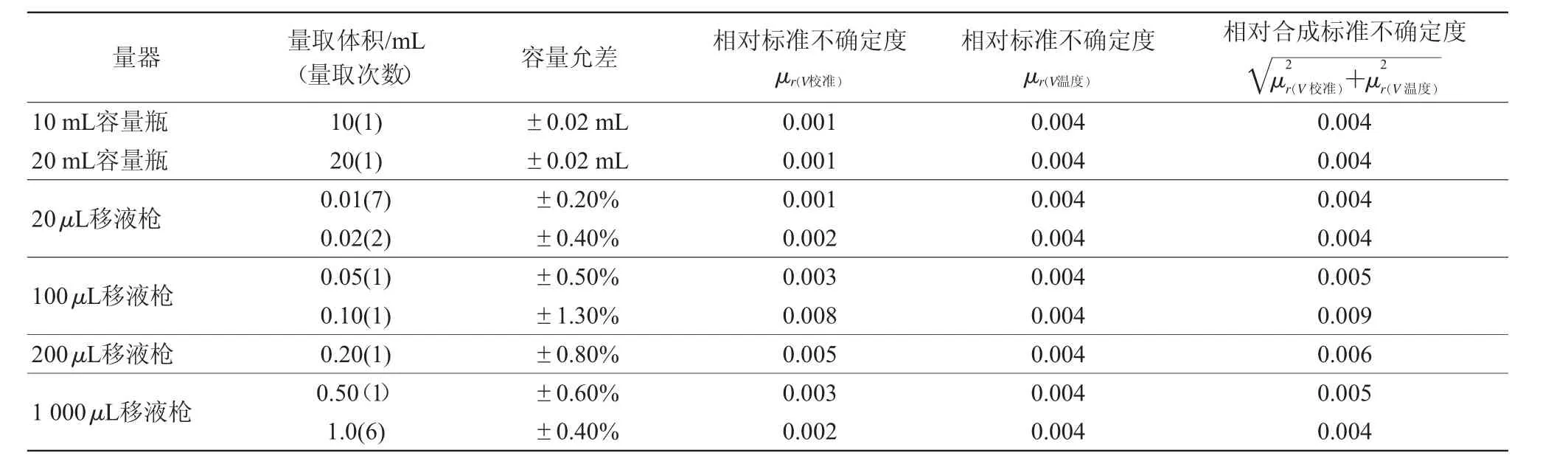
表2 量器引入相对标准不确定度
由此,称量标准物质及配制基质标准曲线时产生的不确定度μr(2.1):

2.2 标准曲线拟合过程产生的不确定度μr(2.2)
配制除虫脲6个质量浓度分别为5.734、11.47、28.67、57.34、114.7、286.7 ng/mL的基质标准曲线进行重复测定3次。由于实验全程加入同一瓶内标使用液,内标浓度一致,无需考虑内标使用液浓度引入的不确定度,只需考虑内标使用体积引入的不确定度,为方便计算实验将内标TPP质量浓度设为1 ng/mL。即除虫脲与内标TPP质量浓度比值x分别为5.734、11.47、28.67、57.34、114.7、286.7,得到除虫脲与内标TPP定量离子丰度比值y,以x为线性横坐标,y为线性纵坐标,用最小二乘法进行线性拟合,得到标准线性方程y=kx+b及相关系数R2,结果详见表3。
基质标准曲线拟合的标准偏差:

基质标准曲线拟合的相对标准不确定度:

式中:n为基质标准工作液测定的点数,n=6;p为样品平行测定的个数,p=6;yi为各基质标准工作液浓度对应的响应比值(Ai目标物/Ai内标);x0为样品测得浓度均值,以回归标准曲线校正所得含量均值计,为97.70 ng/mL(表4);x¯为标准曲线各点浓度的平均值,为84.10 ng/mL(表3);b为直线截距,为0.007 3。
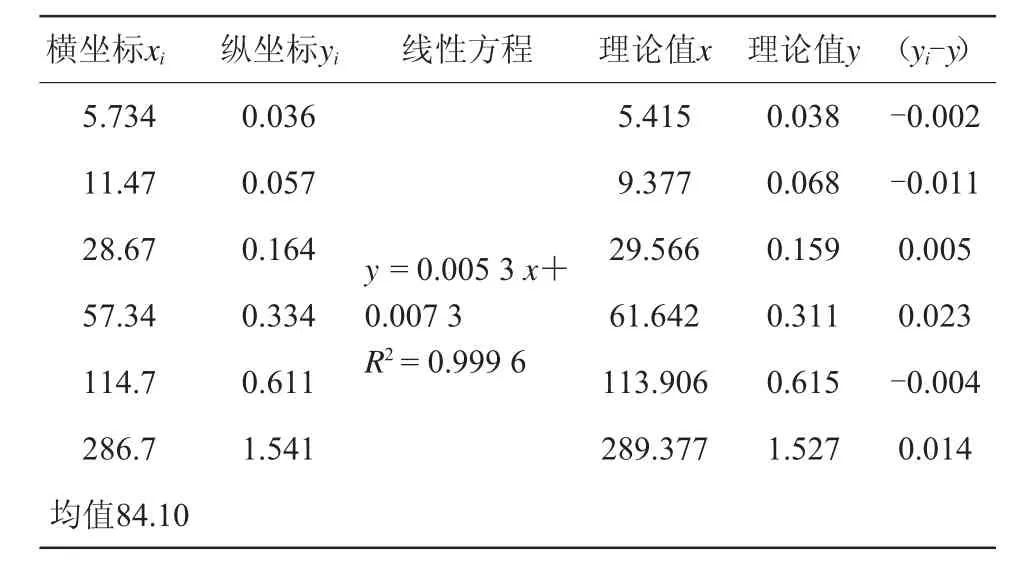
表3 基质标曲拟合数据
注:把xi代入直线方程得理论y值,把yi代入直线方程得理论x值。
因此,基质标曲拟合的相对标准不确定度μr(2.2)=0.012。
2.3 茶叶试样前处理产生的不确定度μr(2.3)
试样称量、稀释时产生的不确定度是未知试样前处理过程常见的不确定度。实验全程加入同一瓶内标使用液,内标浓度一致,由于考虑了内标使用体积产生的不确定度,即试样稀释体积不影响到定量结果,实验无需再考虑试样前处理稀释体积引起的不确定度。
2.3.1 茶叶试样称量产生的不确定度μr(m)
检定天平检定证书,其最大允许误差为0.5 mg,已知称样2 g,取矩形分布,则标准不确定度μ(m)=0.289 mg,相对标准不确定度为μr(m)=0.000 1。
2.3.2 茶叶试样加入内标体积产生的不确定μr(v)
前处理过程在茶叶试样中加入0.1 mL内标TPP使用液,根据移液枪检定证书容量允差及温度波动引起的体积变化(表2),相对标准不确定度μr(v)=0.009。
由此,未知试样前处理时产生的相对标准不确定度μr(2.3):

2.4 茶叶试样测定重复性产生的不确定度μr(2.4)
准确称取6份茶叶试样,经提取、净化步骤,进样分析。试样平行测量结果见表4。

表4 试样测定重复性结果
试样测定均值S¯=0.479 mg/kg(n=6),标准偏差S样品=0.006 2 mg/kg,标准不确定度mg/kg,相对标准不确定度
2.5 茶叶试样加标回收产生的不确定度μr(2.5)
称取阴性基质随行加标,考察回收率产生的不确定度。阴性基质加标回收实验进行6次测定(n=6),除虫脲加标量为114.7 ng,回收率计算结果见表5。

表5 茶叶中除虫脲加标回收率结果
加收率均值S¯=95.2%(n=6),回收率标准偏差S回收=1.4%,标准不确定度相对标准不确定度
使用t检验法检验平均回收率与回收率期望值100%的差异:

查表得,t0.95(5)=2.57<8.00,显示回收率结果与100%有显著性差异,表明样品测定需要用回收率进行结果校正,校正后的结果为0.503 mg/kg。
2.6 不确定度的合成
除虫脲各分量的相对标准不确定度见表6,则除虫脲的相对合成标准不确定度:
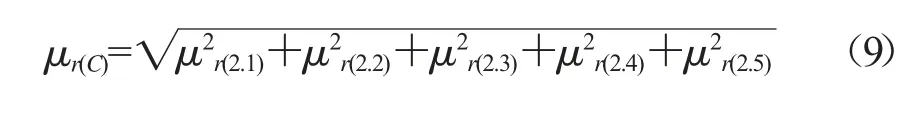

表6 茶叶中除虫脲各分量的相对标准不确定度统计
折算成除虫脲合成标准不确定度为:

2.7 扩展不确定度及结果
在P=95%置信水平,测量结果扩展不确定度取扩展因子k=2,则茶叶中除虫脲残留量测定的扩展不确定度为:U(C)=kμ(C)=0.028 mg/kg。因此,茶叶试样中除虫脲的残留量表示为(0.503±0.028)mg/kg(k=2)。
3 结论与讨论
通过液相色谱-串联质谱法测定茶叶中除虫脲残留量的不确定度,得出该测定方法的扩展不确定度为0.028 mg/kg。从表6可见,称量标准物质及配制基质标准曲线时产生的不确定度μr(2.1)分量对合成不确定度的贡献最大,标准曲线拟合过程产生的不确定度μr(2.2)次之。其中,在合成μr(2.1)众多的小分量中,配制基质标准曲线时引入的不确定度μr(V),影响最为显著。因此,配制基质标准曲线时使用允差范围小的移液枪,保持恒温恒湿的实验环境,注意操作人员的熟练水平,把控好内标加入体积的重复性,皆有助于减小配制基质标准曲线引入的不确定度。此外,尽可能提高曲线拟合的线性,例如标准曲线每个点测量多次,测量值取平均值进行拟合,线性范围内多取几个浓度点,并确保样品测定结果在曲线范围的中高段等,可降低测定结果的不确定度。
