Landscape and Dynamics of the Transcriptional Regulatory Network During Natural Killer Cell Differentiation
2020-09-02KunLiYangWuYoungLiQiaoniYuZhigangTianHaimingWeiKunQu
Kun Li,Yang Wu,Young Li,Qiaoni Yu,Zhigang Tian,2,Haiming Wei,2,*,Kun Qu,2,3,*
1 Department of Oncology,The First Affiliated Hospital of USTC,Division of Molecular Medicine,Hefei National Laboratory for Physical Sciences at Microscale,School of Basic Medical Sciences,Division of Life Sciences and Medicine,University of Science and Technology of China,Hefei 230021,China
2 CAS Center for Excellence in Molecular Cell Sciences,The CAS Key Laboratory of Innate Immunity and Chronic Disease,University of Science and Technology of China,Hefei 230027,China
3 School of Data Science,University of Science and Technology of China,Hefei 230026,China
KEYWORDS NK cell;ATAC-seq;Programmed differentiation;FOSL2;EGR2;Dynamic regulatory network
Abstract Natural killer(NK)cells are essential in controlling cancer and infection.However,little is known about the dynamics of the transcriptional regulatory machinery during NK cell differentiation.In this study,we applied the assay of transposase accessible chromatin with sequencing(ATAC-seq) technique in a home-developed in vitro NK cell differentiation system.Analysis of ATAC-seq data illustrated two distinct transcription factor(TF)clusters that dynamically regulate NK cell differentiation.Moreover,two TFs from the second cluster,FOS-like 2(FOSL2)and early growth response 2 (EGR2),were identified as novel essential TFs that control NK cell maturation and function.Knocking down either of these two TFs significantly impacted NK cell differentiation.Finally,we constructed a genome-wide transcriptional regulatory network that provides a better understanding of the regulatory dynamics during NK cell differentiation.
Introduction
Natural killer (NK) cells are innate lymphocytes that survey the environment and protect the host from infected or cancerous cells.As their name implies,NK cells represent an early line of defense during pathogen invasion by directly killing infected cells and secreting inflammatory cytokines [1].Additionally,NK cell-based immunotherapy has become an emerging force in cancer treatment and will play an essential role in the future treatment [2-6].For instance,adoptive NK cell immunotherapy has become increasingly popular due to its capability to induce graft-versus-leukemia effects without causing graft-versus-host disease in patients [7].Therefore,major efforts are currently underway to fully utilize the antitumor properties of NK cells in the clinic.In addition,a variety of methods have been implemented to expand NK cells,establish a microenvironment that favors NK cell activity,and redirect the activity of NK cells onto tumor cells [8].On the other hand,many researchers use umbilical cord blood(UCB) CD34+cells to produce abundant NK cells [9-18]for clinical application without feeding cells by adding various cytokines [10].In our previous work,we have developed a method to obtain sufficient functional NK cells by simply adding a mixture of cytokines and provide a mechanism by which NK cells can be used to treat leukemia [19].The use of NK cells for immunotherapy relies on the presence of a great number of NK cells with optimal cytotoxic activity[4];therefore,a comprehensive understanding of the regulatory circuits during NK cell differentiation is particularly important for boosting the efficacy of clinical treatments.However,the mechanisms underlying NK cell differentiation are not well understood.Studies have shown that transcriptional factors (TFs) play an essential role in driving NK cell maturation,and many TFs have been well studied in this process [1].Additionally,it is known that different TFs play various roles at distinct stages of differentiation [1].For example,PU.1 is a TF that is known to drive hematopoietic stem cell (HSC) differentiation into the earliest myeloid and lymphoid progenitors [20],whereas T-bet is an essential TF in the control of NK cell maturation and IFN-γ secretion [21].However,how TFs work in concert to enforce the NK cell phenotype is not clear.
Assay of transposase accessible chromatin with sequencing(ATAC-seq),a newly developed epigenomic profiling technique[22],has been widely used to profile the epigenetic landscapes of cells at specific stages of interest and thereby delineates the underlying regulatory mechanisms of gene expression.For example,previous reports using ATAC-seq identified an exhaustion-specific enhancer that regulates PD-1 expression,thereby elucidating the regulatory mechanism of gene expression in exhausted CD8+T cells [23,24].Alternately,ATAC-seq analysis of pure cancer cell populations of human small cell lung cancer (SCLC) identified a novel TF,nuclear factor I B (NFIB),which is necessary and sufficient to drive the metastatic ability of SCLC cells [25].ATAC-seq has also enabled researchers to track the epigenomic state changes in patient-derived immune cells [26],and to survey how the personal regulomes of the cutaneous T cell lymphoma patients determine their responses towards histone deacetylase inhibitor (HDACi) anti-cancer drugs [27].More recently,ATAC-seq was applied to better understand the control of NK cells in innate immune memory during infection[28],illustrating the importance of the topic as well as the power of the technique used in this study.
Here,we have developed a systematic method to characterize the chromatin accessibility and regulatory network dynamics during NK cell differentiation from UCB CD34+HSCs based on ATAC-seq.Motif and enrichment analyses from the Hypergeometric Optimization of Motif EnRichment(HOMER) [29] and Genomica [27] show that many TFs play important roles during NK cell differentiation.By integrating gene expression profiles from our previous study [19],two novel TFs,FOS-like 2 (FOSL2) and early growth response 2(EGR2),were identified to be essential to drive NK cell maturation.
Results
Landscape of DNA accessibility during NK cell differentiation
To elucidate the regulatory networks during NK cell differentiation,we developed a culture procedure to obtain differentiated NK cells from UCB CD34+cells using a cocktail of cytokines[19].The differentiation process took 35 days.Interestingly,after culturing of cord blood stem cells for 3 weeks,the proportion of NK cells rapidly increased from 5% on day 21 to approximately 60% on day 28,and peaked at close to 100%on day 35(Figure 1A,Figure S1A)[19].To elucidate the dynamic changes of transcriptional regulation during NK cell differentiation,we interrogated the landscape of chromatin accessibility using ATAC-seq at 8 different time points,with 2 replicates each along the process (Figure 1B).Multiple bioinformatics analyses(see Materials and methods)were then performed to obtain the differentially accessed chromatin sites,enriched TFs,and genome-wide regulatory elements.At least 50,000 cells were obtained from each sample,resulting in more than 78 million reads on average with a total of 1260 million reads for all time points (Table S1).From this dataset,143,570 accessible DNA sites were identified (P< 10-7,FDR <10-7).Transcription start site(TSS)enrichment score and ATAC-seq signal analysis indicated that the dataset was of high quality with a strong signal-to-background noise ratio and expected fragment length distribution (Figure S1B-E).Moreover,the dynamics of the chromatin accessibility around the known surface marker genes were consistent with changes of their gene expression levels during NK cell differentiation(Figure S1D) [30,31].Pearson correlation analysis on the biological replicates at each time point also showed that our ATAC-seq profiles were highly reproducible (Figure S2A and B).Within all the accessible sites,only a very small portion(6.48%) were conserved across all stages (Figure S3A),while the majority of peaks changed over time,suggesting significant chromatin dynamics during NK cell differentiation.Peaks that were detected at all stages were enriched in Gene Ontology(GO) terms such as RNA transcription and other functions to maintain basic physiological activities(Figure S3B).In contrast,more and more immune response-related GO terms were enriched as the time went by,suggesting the activation of critical genes that govern NK cell function.
Several TFs are known to regulate NK cell differentiation,such as PU.1/Spi-1 proto-oncogene (SPI1) and T-bet/T-box transcription factor 21 (TBX21).The former is a NK cell repressor and the latter an activator.Gene expression profiles from our previous study (GEO:GSE87787) [19] indicated down-regulation of theSPI1gene and up-regulation of theTBX21gene during NK cell differentiation (Figure 1C).The levels of proteins encoded by these two genes were evaluated using flow cytometry,which suggested the same trends (Figure 1D).ATAC-seq analysis revealed four peaks at the intronic enhancers ofSPI1and two peaks at the promoter and intronic enhancer ofTBX21,respectively(Figure 1E).Accessible chromatin sites around theSPI1gene clearly became unavailable and those around theTBX21gene gradually became available during the process,supporting the notion that the epigenetic dynamics of key regulators are consistent with their corresponding gene and protein expression levels.The consistency between the epigenetic and gene expression profiles of TFs known to drive NK cell differentiation,such as GATA binding protein 3 (GATA3) [1] and eomesodermin(EOMES)[1],is shown in Figure S1E,indicating the feasibility of predicting transcriptional regulatory networks from ATACseq profiles.

Figure 1 DNA accessibility during NK c
Epigenomic signatures of different stages during NK cell differentiation
To determine the differences in regulatory DNA activity among different stages during NK cell differentiation,we performed pairwise comparisons of the ATAC-seq signals between the corresponding samples,together with intrinsic analysis[32],a method that highlights the elements with varied accessibility across individuals but not between replicate samples from the same individual (File S1).We discovered 6401 peaks showing differentially accessed DNA sites across the genome,which were categorized into three distinct clusters via unsupervised hierarchical clustering(Figure 2A,Figure S4A and B).Principal component analysis (PCA) of all samples also illustrated three distinct clusters,which is consistent with the time process of NK cell differentiation representing the early,interim,and late stages of the entire process (Figure S4C).GO analysis of these peaks was performed in the Genomic Regions Enrichment of Annotations Tool(GREAT)[33].Cluster I comprises 1584 elements that are more accessible at the early stage (days 7-21) of differentiation.Peaks in cluster I are significantly enriched in the GO terms of metabolic processes that are necessary for cell viability,proliferation,and differentiation (Figure 2B,upper panel;10-8<P<10-4).Cluster II comprises 4461 elements that are highly accessible at the interim and late stages (days 24-35) of NK cell differentiation.GREAT analysis revealed that peaks in cluster II are strongly enriched (P<10-50) in immune-relevant GO terms,such as immune system process,immune response,and immune system development,among others (Figure 2B,lower panel),indicating that the epigenetic states of functional immune cell-specific genes were activated throughout the process.Cluster III consists of only 356 peaks,with no enrichment for GO terms,and were therefore not included for downstream analyses.
We next examined whether the DNA accessibility signatures at different stages are correlated with the expression profiles of corresponding genes.By comparing the ATAC-seq profiles with the genome-wide microarray data during NK cell differentiation,we found that genes that gained chromatin accessibility (cluster II) showed significant increases in expression(Figure 2C,right panel;P<0.001),while genes that lost chromatin accessibility (cluster I) had decreased expression(Figure 2C,left panel;P<0.001),indicating a high correlation between epigenetic and RNA profiles.
Chromatin structures and epigenetic modifications are known to regulate gene expression [34].However,the chronological order of the dynamics of the chromatin accessibility,gene expression,and cell phenotype has not yet been well studied.Here,we integrated the ATAC-seq signals,microarray profile,and percentage of NK cell counts to examine the temporal dynamics of these three features.Interestingly,we noticed that the accessibility of mature NK cell-specific peaks(cluster II in Figure 2A) started to increase on day 14,the expression of NK cell-specific genes was turned on approximately two days later,while the percentage of NK cell counts started to increase after day 21(Figure 2D).These results suggest a clear chronological order of the changes in chromatin structure,gene expression,and cell population during NK cell differentiation.
TF occupancy network during NK cell differentiation
TFs bind to cognate DNA sequences with patterns termed motifs.Therefore,we could predict TF occupancy on chromatin using DNA accessibility data from ATAC-seq for constructing regulatory networks [26].To identify potential drivers of NK cell differentiation,we screened TFs that were enriched at accessible elements in clusters I and II using HOMER in the search modes with default (Figure 3A) or setting background (Figure S5A).Our finding suggests that NK cell differentiation and maturation require a variety of TFs.TFs enriched at cluster I peaks are potential regulators at the early stage of NK cell differentiation,while those enriched at cluster II peaks could be critical regulators at the interrim and late stages.We found several TF families that were significantly enriched (P<10-10).Many of them are known to be important for NK cell differentiation,such as the Runt-related transcription factor (RUNX) families,ETS proto-oncogene(ETS) families [20,35],and CCAAT enhancer-binding protein(CEBP) families [36],supporting the reliability of this method to detect critical regulators.For instance,PU.1 is widely expressed and controls multiple stages of bone marrow and lymphocyte differentiation in a variety of hematopoieticderived lineages [37-41].An decrease in the number of NKcell progenitor(NKP)and immature NK(iNK)cells has been detected in chimeric mice [20],indicating that PU.1 may play an important role during the early stage of NK cell differentiation.Several known TFs,such as RUNX [35],E2A immunoglobulin enhancer binding factors E12/E47 (E2A)[42],T-bet[21],signal transducer and activator of transcription 5 (STAT5) [43],and EOMES [21],are also enriched at the interrim and late stages.The most enriched TFs at cluster II peaks belong to the RUNX family:RUNX1,RUNX,and RUNX2(Figure S5A),which are key regulators of lymphocyte lineage-specific gene expression [44].
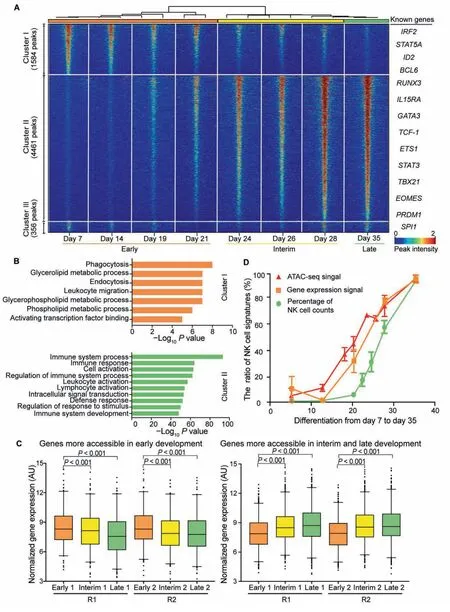
Figure 2 Differential epigenetic regulation elements during NK cell differentiation
Motif search of differential ATAC-seq peaks could provide information about the transcriptional regulatory network.However,one caveat of this method is the inability to distinguish similar binding motifs of TF family members.Thus,we sought to apply the Genomica’s module map algorithm and ‘‘TF footprint”analysis to better predict TF occupancy on accessible sites by integrating the ATAC-seq and gene expression microarray profiles (Figure 3).
In total,242 vertebrate TF motifs were obtained from the JASPAR database [45].We then used Genomica [27] to produce a time point-specific TF occupancy network(Figure 3B).This analysis revealed distinct patterns of TF accessing to DNA at different time points.Many known TFs were enriched,such as STAT5,which is an IL-15 downstream signaling molecule and is indispensable throughout the lifetime of NK cells.Deficiency of STAT5a/b has been reported to result in complete elimination of NK cells,demonstrating the important and non-redundant effects of STAT5 [43].The JAK-STAT pathway has also been shown to be an important signaling pathway used by various cytokines and growth factors [46].The interferon regulatory factor (IRF) family regulates a variety of processes,including hematopoietic development,tumorigenesis,host immunization,and pathogen defense[47,48].IRF2 is required to maintain the normal differentiation of NK cells in a cell-intrinsic manner [49,50],and IRF2-deficient NK cells show reduced levels of maturation markers and IFN-γ production during stimulation [49,50].Tbet and EOMES are members of the T-box family and are known to control different aspects of NK cell differentiation and maturation [21,51,52].
In addition,several novel TFs were also enriched,such as FOSL2 and EGR2,suggeting that they are potential regulators of NK cell differentiation.These TFs were assessed along with the other enriched TF families from the motif analysis to identify which family members were expressed or differentially expressed during NK cell differentiation to further filter out true regulators.At each time point,we plotted both the expression value (colored from red to green to represent high to low expression in the gene microarray profile)and the motif enrichment score (shown by the circle size representing the -logPvalue of the enrichment) in the same figure (Figure 3C).We observed that known regulators were highly expressed and enriched at different stages,same as FOSL2 and EGR2.Through the integral analyses (Figure 3A-C),we predict FOSL2 and EGR2 as potential regulators.
DNA sequences that are directly occupied by DNA-binding proteins are protected from transposition,and therefore the resulting sequence ‘‘footprint”reveals the presence of a DNA-binding protein at each site,analogous to DNase digestion footprints.The TF‘‘footprint”analysis of our ATAC-seq profile provided further evidence of direct occupancy of a TF candidate on genomic DNA and thus refined the prediction of potential regulators.We illustrated the ‘‘footprints”of two known regulators,STAT5 and T-bet,and observed higher DNA accessibility and deeper ‘‘footprints”flanking their motifs in the interim and late stages than those in the early stage during NK cell differentiation (Figure 3D).Similarly,‘‘footprints”of the TFs FOSL2 and EGR2 were also deeper and more accessible at the interim and late stages.These data suggest that not only the binding motifs of these two TFs are enriched at stage-specific peaks,but also these TFs most likely physically bind to these accessible chromatin sites,indicating that they are functional regulators of NK cell differentiation(Figure 3D).Overall,the results from footprint analysis agree with those from the HOMER and Genomica’s motif enrichment analyses.
Genes that are regulated by any TF of interest can be predicted by combining the TF motif and ‘‘footprint”analyses.We thus predicted the genes regulated by TFs including RUNX,T-bet,FOSL2,and EGR2,and integrated the gene expression profiles at each time point (Figure S5B;Table S2).We found that genes regulated by each of these TFs were also highly expressed at the interim and late stages and were significantly enriched in GO terms of immune system construction and other related functions.TF ETS protooncogene 1 (ETS1) was reported to drive early stages of NK cell differentiation [53],and we found that there was a FOSL2-binding site in a dynamically accessible site on the gene body ofETS1,suggesting FOSL2 might regulate NK cell differentiation throughETS1(Figure 3E,left panel).TF GATA3 was found to regulate liver-specific NK cells,IFN-γ production,and T-bet expression in mice [54].Similarly,we found that both FOSL2 and EGR2 bind to the gene body ofGATA3(Figure 3E,right panel),suggesting that the TFs FOSL2 and EGR2 may also regulate NK cell differentiation through regulation ofGATA3.

Figure 3 TFs enriched at different stages of NK cell differentiation
FOSL2 and EGR2 affect NK cell differentiation
FOSL2 belongs to the Fos gene family,which encodes a leucine zipper protein and forms the AP-1 TF complex by dimerization with the JUN family.Thus,FOS proteins have been suggested as key regulators of transformation,differentiation,and cell proliferation.The GO annotations ofFOSL2include sequence-specific DNA binding,TF activity,and RNA polymerase II specific DNA binding.A previous report has shown that FOSL2 is constitutively expressed in adult T-cell leukemia(ATL),up-regulatesCCR4expression,and promotes ATL cell growth,together with JunD proto-oncogene,AP-1 transcription factor subunit(JUND)[55].EGR2encodes a protein that contains three tandem C2H2-type zinc fingers.GO annotations of this gene include ligase activity,sequence-specific DNA binding,and TF activity.Previous reports have shown that EGR2 regulate T-cell and B-cell function in homeostasis and adaptive immune responses by controlling inflammation and promoting antigen receptor signaling [56,57].
Since their regulatory functions in NK cell differentiation have not been well characterized,we performed loss-offunction experiments to assess the effects ofFOSL2andEGR2on NK cell differentiation.We first validated their expression levels with real-time PCR,and found that their expression gradually increased in later stages of NK cell differentiation,consistent with our bioinformatics analysis (Figure S6A).Subsequently,we infected the cultured cells with TF-specific shRNA-and control shRNA-containing lentiviruses,which are represented by GFP expression (Fig-ure 4A).We then sorted the GFP-positive cells and observed dramatically reduced expression levels of FOSL2 and EGR2(Figure 4B),suggesting positive targeting of the TF-specific shRNAs.During NK cell differentiation,we observed a nearly 30% reduction in the proportion of differentiated NK cells in GFP-positive cells at day 28 and day 35 (Figure 4C and D).However,in the non-successfully transfected (GFP-negative)cells,the proportion of differentiated NK cells was not affected at the same time point (Figure 4E and F).These results indicate that knockdown ofFOSL2andEGR2expression,but not viral infection,inhibits NK cell differentiation,suggesting that FOSL2 and EGR2 are necessary for NK cell differentiation.We then tested another important marker of NK cell maturation CD11b [1],and found that it was significantly reduced in GFP-positive NK cells,suggesting that FOSL2 and EGR2 might affect NK cell maturation (Figure 4G).Overall,we predicted these two TFs FOSL2 and EGR2 as key regulators based on ATAC-seq and gene microarray profile analyses,and then experimentally verified that they indeed affect NK cell differentiation.
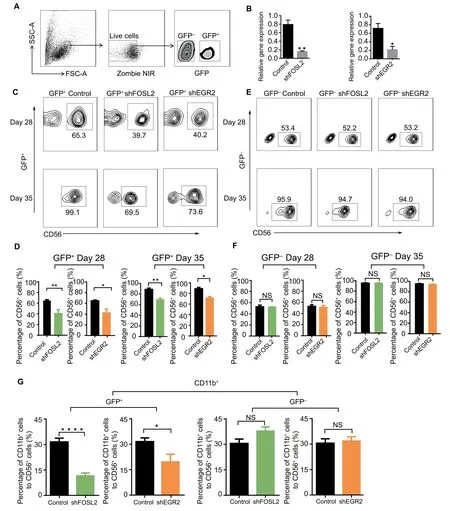
Figure 4 Function of EGR2 and FOSL2 during NK cell differentiation
In addition,we attempted to identify the signaling pathways through which FOSL2 and EGR2 were involved in driving NK cell differentiation by performing module map analysis of all the differential peaks.Eighteen modules were identified according to the patterns of their accessibility (Table S3),several of which were significantly enriched with known TFs that regulate NK cell differentiation (Figure S6B),includingFOSL2andEGR2,and several other genes from the JAKSTAT pathway,such as theSTATfamily,IRF2,TBX21,andGATA3.These results suggested that FOSL2 and EGR2 may regulate NK cell differentiation through the JAK-STAT pathway (Figure S6C).
Transcriptional regulatory network dynamics during NK cell differentiation
The dramatic chromatin accessibility differences during NK cell differentiation prompted us to check the time pointspecific transcriptional regulatory network.Although some TFs have been discovered to regulate NK cell differentiation,little is known about the dynamics of the entire regulatory network during this process.Since the TF footprint pattern from the ATAC-seq reads could simultaneously and directly reveal the binding profiles of hundreds of TFs with known cognate motifs [22],together with gene expression profiles,we could construct a regulatory network at each time point and assess how it changes during NK cell differentiation.First,we used HOMER to identify enriched TFs that bind to the cluster I and cluster II peaks shown in Figure 3A (P<0.05).We then examined the gene expression profiles of these TFs and found that 120 TFs were expressed at least one stage during the differentiated process.By applying differential analysis,we obtained 14-26 TFs that were distinctly expressed at each stage with fold change(FC) >1.5(Figure S7),and defined them as the nodes of the regulatory network.The connections (edges)between any two TFs were defined as follows:if the TF A’s motif is on the promoter of TF B,then we say TF A regulates TF B and draw an arrow from TF A to TF B.Here,only TFs that were expressed at the specific time point were considered[58].Using this method,we constructed the transcriptional regulatory network at each time point with both the enrichment(Pvalue) and expression information for all the relevant TFs(Figure 5A-E).Interestingly,intensive interconnections appeared for day 7-specific TFs,and quickly vanished after two weeks (Figure 5A).In contrast,the day 35-specific network gradually grew through the induction of relevant TFs.Many known regulators,such as EOMES,TBX21,ETS1,PR/SET domain 1(PRDM1),and GATA3,as well as FOLS2 and EGR2,were increasingly enriched in the network (Figure 5E).Similar phenomena were also observed on other networks (Figure 5B-D).The dynamics of the transcriptional regulatory networks seem to explain the increase in the proportion and the differentiation of NK cells.
Discussion
NK cells are important innate immune cells that have been recognized as efficient effector cells to treat tumors.To better understand the differentiation machinery of NK cells and to identify new regulators in the process,we studied the landscape of active elements during NK cell differentiation using the sensitive ATAC-seq method.As a result,we find three distinct clusters of DNA accessible elements.Further analysis shows that the chromatin accessibility is correlated well with the expression levels of the corresponding genes.In short,this study provides an epigenomic landscape and dynamics of NK cell differentiation and presents fundamental profiles for studying the relationship between chromatin accessibility,gene expression,and cell growth during this process (Figure 2).
TFs bind to their motifs and are often obligate nucleosome evictors,and therefore we can use ATAC-seq to predict critical regulators in NK cell differentiation [26].By motif analysis in HOMER,we find that several known TFs are enriched at different stages during NK cell differentiation.Similar results have also been obtained using Genomica(Figure 3B).The discovery of known regulators strongly supports the reliability of our analysis.Furthermore,by integrating results from HOMER,Genomica,and motif footprint analysis,we have identified two novel TFs,FOSL2 and EGR2,which are essential for NK cell maturation.Knockdown of either of these two TFs significantly inhibits NK cell transformation in thein vitroNK cell differentiation system.Module map analysis suggests that these two TFs may regulate NK cell differentiation through the JAK-STAT pathway,and therefore further studies of this pathway may facilitate the generation of NK cells and thus promote the NK cell-based immunotherapy.Overall,this study also provides a framework to identify new regulators from chromatin accessible data for NK cell differentiation.

Figure 5 Transcriptional regulatory network during NK cell differentiation
TFs do not usually function alone,they always interact with other molecules to fulfill their unique roles.Hence,we have depicted the transcriptional regulatory networks at different stages during NK cell differentiation.In order to construct a stable transcriptional regulatory network,we have performed a rigorous screening of TFs to avoid stochastic fluctuations,and integrated both the enrichment (Pvalue) and expression information for all the relevant TFs.Therefore,although alterations in either the enrichment or gene expression cutoff may result in different networks,the most critical TFs to the regulatory process still remain.However,since the screening of TFs mainly rely on the gene microarray,which is not as accurate as RNA-seq,the structure of these predicted networks may not be very robust.
From our previous study,we notice that with a minimal cytokine cocktail,we can generate sufficient number of functional NK cells that express the cytokines necessary for NK cells and have a high effect on tumor cells[19].Although there may also be a certain proportion of other lymphocytes,in vitroexpansion of NK cells from peripheral blood (PB) or UCB cells has been successfully performed and developed for several clinical strategies to treat cancers [4,5,59-61].Therefore,a comprehensive understanding of the regulatory patterns at each differentiation stage of thein vitro-derived NK cells in this system will help to uncover the underlying mechanisms of NK cell differentiation.The transcriptional regulatory networks revealed in this study will lay the foundation for efficientin vitroproduction of effective NK cells,thus facilitating NK cell-based immunotherapy.Moreover,we have identified two novel TFs,FOSL2 and EGR2,as essential regulators in controlling NK cell maturation and function.We have also predicted the potential signaling pathways in which these two TFs may be involved and have illustrated the dynamics of the transcriptional regulatory networks during NK cell differentiation.In spite of the advantages of our strategy,there are two main limitations of this study.1) Although induced NK cells are very similar to those producedin vivo,these two types of NK cells are not identical.We have observed certain differences between the induced NK cells and the NK cells producedin vivoin terms of chromatin accessibility (data not shown).2) Before NK cells are fully developed,there is always a mixture of cell populations with stem cells,NKP cells,iNK cells,mature NK cells,and other cell types that we are unable to delineate at this moment,since any bulk cell-based analysis may neglect the huge heterogeneity between cells by default.Therefore,to fully uncover the regulatory mechanism,single cell technologies are required in the future to further delineate the cell-to-cell heterogeneity and regulatory dynamics at the single cell precision.
Materials and methods
Samples
UCB samples were collected at birth from women with normal,full-term deliveries at Anhui Provincial Hospital,Hefei,China,after receiving their written informed consent.The culture procedure for NK cell differentiation from UCB CD34+cells has been previously described [19].
Immunofluorescence staining
The cultured cells were post-fixed in 4% paraformaldehyde,blocked with 10% goat serum (Catalog No.31872;ThermoFisher Scientific,Grand Island,NY),before incubating with primary antibodies at 4°C overnight.Secondary antibodies were then added and incubated at 37 °C for 1 h after washes.Cell nuclei were stained with DAPI for 5 min at room temperature.Confocal images were acquired using a Zeiss LS710 microscope.
RNA isolation and real-time PCR
Cultured cells were lysed in TRIzol reagent (Catalog No.15 596026;ThermoFisher Scientific),and total RNA was extracted following the manufacturer’s instructions.cDNA was synthesized with Moloney murine leukemia virus reverse transcriptase(Catalog No.28 025013;ThermoFisher Scientific)and oligo (dT) 20 primers.Then,SYBR Premix Ex Taq (Catalog No.RR420L;TaKaRa,Dalian,China)was used for realtime PCR using primers with sequences listed in Table S4.The data were analyzed using the 2-ΔΔCtmethod.
Lentivirus production and transduction
To produce lentiviral particles,293 T cells were transfected with the plasmids PLKO.1,pRRE,pREV,or pVSV-G using Lipofectamine 2000 (Catalog No.11668-019;Invitrogen,Grand Island,NY) following the manufacturer’s protocol.Then,we harvested the supernatants 48 h and 72 h posttransfection.To remove cell debris,supernatants were centrifuged at 3000 rpm for 10 min,and then,the lentivirus particles were concentrated by ultracentrifugation at 50,000gfor 2 h at 4°C.Finally,the virus particles were gently resuspended in Hanks’balanced salt solution(HBSS)and stored at-80°C.After UCB CD34+cells were cultured with multiple cytokines for 14-18 days,we incubated the lentiviruses and the cultured cells with polybrene(Catalog No.H9268-10G;Sigma Aldrich,Shanghai,China) (5 μg/ml) for 30 min and the mixture was subjected to centrifugation at 1000 rpm for 70 min at 10 °C.
Statistical analysis
Statistical significance was analyzed using unpaired two-tailed Student’st-test or one-way ANOVA.Pvalues less than 0.05 were considered statistically significant.
ATAC-seq and analysis
ATAC-seq was performed as previously described [22],and 2 × 150 paired-end sequencing was performed on an Illumina HiSeq X-10 to yield,on average,78 M reads/sample.Sample reads from biological replicates were then grouped together and divided into eight categories:day 7,day 14,day 19,day 21,day 24,day 26,day 28,and day 35.Intrinsic analysis and other ATAC-seq analysis was performed same as our previous work (see File S1) [26,62].
Differential analysis
All samples were grouped into eight categories (16 samples):day 7,day 14,day 19,day 21,day 24,day 26,day 28,and day 35.Data normalization and significance analysis were performed via pairwise comparison between the eight categories using DESeq2 [63] withP< 0.01,log2FC > 5,and FDR <0.01,and intrinsic analysis [26] with a z-score >1.We finally obtained 6401 differential peaks.Unsupervised clustering was performed using Cluster 3.0 and visualized in Treeview.GREAT[33]was used to predict enriched functions and GO terms.
Identification of stage-specific peaks
As shown in Figure S3,each stage (e.g.,day 7) consists of genes that were more highly expressed(FC >1.5)at this stage compared with all the other stages,and we defined these genes as stage-specific genes.Similarly,we defined peaks that were more accessible (FC >1.5) in one stage compare with all the other stages as stage-specific peaks.Peaks that were accessible at all stages were defined as conservative peaks.
TF motif enrichment analysis
HOMER[29]was used to perform the TF enrichment analysis with the following options:findmotifs.pl input.fa fasta output/(Figure 3A)and findmotifs.pl input.fa human uotputdir-fasta bg.fa (Figure S5A).TF enrichment analysis was performed in Genomica.TF‘‘footprint”analysis was performed in the same way as described in our previous work [27].
Gene module and STRING analysis
Gene module analyses were performed using WGCNA [64]with the options SoftPower=20,minModuleSize=30.Protein-protein interaction analyses were performed using STRING [65].
Construction of transcriptional regulatory networks
We used HOMER to find the TFs that bind to cluster I and cluster II peaks and obtained TFs that could regulate stage differentially expressed elements (P<0.05).We define the region 2 kb upstream of the transcription start site as a promoter.If TF A bound to the promoter of TF B,we defined TF A as a regulator of TF B and then constructed a transcriptional regulatory network.Networks of TFs were assembled from TFs that were expressed in at least one sample.An edge between TF A and TF B indicated that TF A binds to the promoter of TF B [58].
Ethical statement
This study was approved by the Ethics Committee of the University of Science and Technology of China.
Data availability
The raw ATAC-seq data have been deposited in the Genome Sequence Archive[66]at the National Genomics Data Center,Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation (GSA:CRA000846) and are publicly accessible at http://bigd.big.ac.cn/gsa.
CRediT author statement
Kun Li:Conceptualization,Methodology,Software,Writing -original draft,Visualization.Yang Wu:Conceptualization,Data curation,Validation,Writing -original draft.Young Li:Data curation,Validation.Qiaoni Yu:Methodology.Zhigang Tian:Supervision.Haiming Wei:Conceptualization,Writing -review & editing.Kun Qu:Conceptualization,Writing -review & editing,Visualization,Project administration,Funding acquisition.All authors read and approved the final manuscript.
Competing interests
All authors have no competing financial interests.
Acknowledgments
This work was supported by the National Key R& D Program of China (Grant No.2017YFA0102900) to KQ,the National Natural Science Foundation of China (Grant Nos.91640113,91940306,31771428,and 31970858 to KQ,as well as 81330071 to HW).It was also supported by the Fundamental Research Funds for the Central Universities,China (Grant Nos.YD2070002019,WK2070000158,and WK9110000141)to KQ.We thank the USTC Supercomputing Center and the School of Life Science Bioinformatics Center for providing supercomputing resources for this project.We thank the CAS interdisciplinary innovation team for helpful discussion.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2020.12.003.
ORCID
0000-0001-6550-4937 (Kun Li)
0000-0002-4430-428X (Yang Wu)
0000-0002-6595-2577 (Young Li)
0000-0003-3272-7119 (Qiaoni Yu)
0000-0003-2742-2361 (Zhigang Tian)
0000-0002-1675-6502 (Haiming Wei)
0000-0002-5555-8437 (Kun Qu)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- PIMD:An Integrative Approach for DrugRepositioning Using Multiple Characterization Fusion
- SeSaMe PS Function:Functional Analysis of the Whole Metagenome Sequencing Data of the Arbuscular Mycorrhizal Fungi
- SeSaMe:Metagenome Sequence Classification of Arbuscular Mycorrhizal Fungi-associated Microorganisms
- iLBE for Computational Identification of Linear B-cell Epitopes by Integrating Sequence and Evolutionary Features
- GTB-PPI:Predict Protein-protein Interactions Based on L1-regularized Logistic Regression and Gradient Tree Boosting
- Exploring Potential Signals of Selection forDisordered Residues in Prokaryotic and Eukaryotic Proteins