SeSaMe:Metagenome Sequence Classification of Arbuscular Mycorrhizal Fungi-associated Microorganisms
2020-09-02JeeEunKangAntonioCiampiMohamedHijri
Jee Eun Kang * ,Antonio Ciampi ,Mohamed Hijri *
1 Institut de Recherche en Biologie Ve´ge´tale,De´partement de Sciences Biologiques,Universite´ de Montre´al,Montre´al,QC H1X 2B2,Canada
2 Department of Epidemiology,Biostatistics and Occupational Health,McGill University,Montre´al,QC H3A 1A2,Canada
KEYWORDS SeSaMe;Spore-associated symbiotic microbes;Arbuscular mycorrhizalfungi;Taxonomic classification;3-codon DNA 9-mer
Abstract Arbuscular mycorrhizal fungi(AMF)are plant root symbionts that play key roles in plant growth and soil fertility.They are obligate biotrophic fungi that form coenocytic multinucleated hyphae and spores.Numerous studies have shown that diverse microorganisms live on the surface of and inside their mycelia,resulting in a metagenome when whole-genome sequencing(WGS)data are obtained from sequencing AMF cultivated in vivo.The metagenome contains not only the AMF sequences,but also those from associated microorganisms.In this study,we introduce a novel bioinformatics program, Spore-associated Symbiotic Microbes (SeSaMe),designed for taxonomic classification of short sequences obtained by next-generation DNA sequencing.A genus-specific usage bias database was created based on amino acid usage and codon usage of a three consecutive codon DNA 9-mer encoding an amino acid trimer in a protein secondary structure.The program distinguishes between coding sequence (CDS)and non-CDS,and classifies a query sequence into a genus group out of 54 genera used as reference.The mean percentages of correct predictions of the CDS and the non-CDS test sets at the genus level were 71% and 50% for bacteria,68% and 73%for fungi (excluding AMF),and 49% and 72% for AMF (Rhizophagus irregularis),respectively.SeSaMe provides not only a means for estimating taxonomic diversity and abundance but also the gene reservoir of the reference taxonomic groups associated with AMF.Therefore,it enables users to study the symbiotic roles of associated microorganisms.It can also be applicable to other microorganisms as well as soil metagenomes.SeSaMe is freely available at www.fungalsesame.org.
Introduction
Arbuscular mycorrhizal fungi(AMF)are plant root inhabiting fungi,of the subphylum Glomeromycotina,which form symbioses with more than 80% of vascular plants worldwide [1].They supply plants with essential nutrients particularly phosphorus and nitrogen,protect them against soil-borne pathogens,and alleviate their abiotic stresses [1-3].Therefore,AMF-based inoculants have been applied in agriculture as a biofertilizer and in phytoremediation for cleaning up contaminated soil [2,4-7].Despite the ecological,agricultural,and environmental importance of AMF,their genetics is poorly understood due to their complex genome organization.They form coenocytic hyphae,reproduce through multinucleated asexual spores,and are strict symbionts[8].Furthermore,it is suggested that AMF are heterokaryons,although this is under debate [9].In addition,numerous studies reported that bacteria and fungi inhabit the surface and the interior of mycelia and spores[10-14].In 2012 and 2013,Tisserant et al.[15,16]separately published the transcriptome and genome of the AMFRhizophagus irregulariscultivatedin vitro.However,only a few AMF taxa are able to grow in axenicin vitrosystems with transformed roots as a host.Thus,whole-genome sequencing (WGS) data from AMF spore DNA originating fromin vivocultures (a conventional cultivation method in a pot culture with a host plant),contain a substantial number of non-AMF DNA sequences,but do provide important information on the microbial communities associated with AMF.In contrast,WGS data fromin vitropetri-dishes contain fewer non-AMF sequences,because antibiotics are used to initiate axenic cultures [17].
Taxonomic classification of WGS data obtained from AMF cultivatedin vivousing current bioinformatics approaches is challenging because these data represent a complex metagenome containing sequences of prokaryotic and eukaryotic microorganisms.Two major approaches for taxonomic classification of random whole metagenome sequencing data (e.g.,whole metagenome shotgun sequencing data)include composition-based methods and similarity-based search methods [18,19].The latter ones include BLAST and its sister programs that are adequate for inferring functions of a query sequence[19,20].Nevertheless,they have limitations in taxonomic classification,because they calculate scores based on a 20 by 20 matrix containing the overall rates of the 20 amino acid substitutions created from the most conserved regions of proteins.The same matrix is applied to all types of query sequences,irrespective of functions,structures,and taxonomic groups.However,due to a lack of bioinformatics tools for analyzing random whole metagenome sequencing data,similarity-based search methods have been commonly used for taxonomic classification.In addition to similaritybased search methods,taxonomic classification pipelines for analyzing targeted metagenome sequencing data (e.g.,16S rRNA gene-based metagenome sequencing data) have been widely used for analyzing random whole metagenome sequencing data in combination with homology search program.Numerous repository databases and pipelines have been developed based on the 16S rRNA gene.However,a previous study has reported the horizontal gene transfer of 16S rRNA genes in prokaryotic organisms and the multiple heterogeneous rRNA genes within a single prokaryotic cell [21].Therefore,they may cause misrepresentation of data if they are not properly dealt with,which may result in erroneous taxonomic classification.
Composition-based methods utilize unique sequence properties such as codon usage bias,compositional patterns in nucleotide sequences(k-mers),and GC content that have been widely used for studying microbial genome evolution in areas of bioinformatics [18,22-26].K-mers are subsequences of length k in a DNA sequence (e.g.,tetramer or 4-mer:ATGT).Composition-based methods using k-mers have been employed in bioinformatics programs for taxonomic classification of random whole metagenome sequencing data [27].They have a number of advantages over similarity-based search methods.It is estimated that more than 99%of existing microorganisms cannot be cultured in laboratory conditions[28],and microbial sequences available in bioinformatics databases represent only a tiny fraction of the diversity of existing microorganisms.Therefore,composition-based methods,which do not require sequence alignments but make predictions based on microorganism’s unique sequence signatures,supposedly excel in taxonomical classification of novel sequences.However,existing bioinformatics programs based on composition-based methods are designed for prokaryotic organisms and their utilization in fungi is inefficient.
In this study,we introduce a novel bioinformatics program for random whole metagenome sequence classification,SeSaMe (Spore-associated Symbiotic Microbes).It provides a means for estimating taxonomic diversity and abundance,as well as,the reservoir of genes of reference taxonomic groups in AMF metagenome.It therefore enables users to study symbiotic roles of taxonomic groups associated with AMF.In order to filter complex evolutionary signals and obtain comparable evolutionary footprints,we calculated codon usage bias based on the amino acid usage and the codon usage of a 3-codon DNA 9-mer that encodes three consecutive amino acids located in a protein secondary structure.We joined three consecutive codons into one unit,and calculated the unit’s relative frequency among synonymous 3-codon DNA 9-mers,which will be hereafter referred to as 3-codon usage.3-codon usage has higher resolution than mono codon usage in assessing the differences among taxonomic groups because evolutionary forces acting on a codon and its encoded amino acid vary widely across protein secondary structures as well as across taxonomic groups.For example,the evolutionary forces acting on the codon AAA,encoding the amino acid Lysine (K) in TGGAAAGTG (WKV),have been different from the evolutionary forces acting on the codon AAA in GACAAAGAA(DKE).We found that 3-codon usage of a 3-codon DNA 9-mer belonging to a protein secondary structure is a taxonomically unique sequence property.SeSaMe calculates a score based on six sets of 3-codon DNA 9-mers from all reading frames (Figure 1),and distinguishes between coding sequence(CDS) and non-CDS.It has an advantage over existing composition-based methods that do not identify nucleotide subsequences with structural roles,or do not consider the biological importance of codons and reading frames.SeSaMe is freely available at www.fungalsesame.org.

Figure 1 Unique advantage of SeSaMe over existing programs
Method
Bacterial and fungal sequence databases
We selected bacterial genera that were dominant in soil based on a literature review [10,28,29-32].While NCBI offered a broad selection of more than 2300 completely sequenced bacterial genomes,we did not have many choices for the majority of fungal phyla.Most of the completely sequenced fungal genomes in NCBI or JGI were Dikarya,while we needed diverse fungal genomes covering Mucoromycotina,AMF,Blastocladiomycota,Neocallimastigomycota,Microsporidia,and Chytridiomycota.We assigned the completely sequenced genomes of 444 bacteria and 11 fungi,includingR.irregularis,to 45 bacterial and 9 fungal genera,respectively,and created CDS and non-CDS databases per genus based on CDS lists provided by NCBI,JGI,and Tisserant et al.[16].The number of genomes per genus varied from 1 to 81,depending on their availability in public databases.The total number of the bacterial genes per genus and the total number of the fungal genes and introns per genus are shown in Tables S1 and S2,respectively.Sequences with an ambiguous nucleotide or with a length shorter than nine—the minimum length of nucleotides required for a 3-codon DNA 9-mer—were excluded.Cryptococcusand Agaricomycetes (Phanerochaete,Scleroderma,andSebacina) belong to the same subdivision,Agaricomycotina,and were grouped together in order to simplify the analysis.
Database design
For selecting a parameter k of k-mer,we chose 3-codon DNA 9-mer as the length of amino acids and of nucleotides,considering the approximate number of amino acids required to form a helical turn in helix and a beta-strand.The program consists of two main components:databases and scoring methods.The major distinguishing feature is the trimer reference sequence database (Trimer Ref.DB).126,093 Protein Data Bank(PDB)entry files were processed with in-house developed parsing programs to extract 7674 amino acid trimers (A.A.Trimers),subunits of protein secondary structures,which were assigned to the sequence variable — A.A.Trimer [33].224,383 3-codon DNA 9-mers,encoding 7674 A.A.Trimers,were assigned to the sequence variable — 3-codon DNA 9-mer.In Trimer Ref.DB,the sequence variables,A.A.Char Trimer (amino acid characteristic trimer),A.A.Trimer,and 3-codon DNA 9-mer,form a three-level hierarchy where A.A.Char Trimer is the highest level (Figure 2).To create A.A.Char,first,we assigned amino acids with similar properties into one group according to polarity and charge of their side chain,and secondly subdivided each group according to their volume(Table 1).Cysteine,glycine,histidine,methionine,and proline have special properties.Therefore,each of them was assigned as a sole member of an A.A.Char group.Generally,multiple A.A.Trimers with similar properties belong to one A.A.Char Trimer.An A.A.Char Trimer and an A.A.Trimer have an A.A.Trimer table and a 3-codon DNA 9-mer table containing multiple members,respectively (Figure 2).
Genus-specific usage bias database (Genus-specific DB)contains the main numerical variable—trimer usage bias.Trimer usage bias is calculated by multiplying the A.A.Trimer usage of A.A.Trimer by the 3-codon usage of 3-codon DNA 9-mer in Trimer Ref.DB (Figure 2).There are 54 CDS Genus-specific DBs and the same number of non-CDS Genus-specific DBs in the program.Each CDS Genusspecific DB contains 1296 A.A.Trimer Usage Tables and 7674 3-codon Usage Tables created based on the CDS database.Non-CDS Genus-specific DB contains the same number of tables derived from the same sequence variables of the Trimer Ref.DB for cost-effective CDS and non-CDS classification.Because SeSaMe compares frequency information of 54 genera calculated based on the same standard genetic code table for the same 3-codon DNA 9-mers,inaccuracy in calculating trimer usage bias of non-CDS is assumed to be insignificant.
Scoring methods
We developed two scoring methods,and each equipped with aPvalue scoring method.The trimer usage probability scoring method classifies a query sequence into one out of 54 genus references,while the rank probability scoring method classifies a query sequence into one out of 13 taxonomic groups:Clostridia,Bacilli,Oscillatoriophycideae,Nostocales,Acidobacteriales,Betaproteobacteria,Deltaproteobacteria,Gammaproteobacteria,Alphaproteobacteria,Actinobacteria,AMF (R.irregularis),Agaricomycotina,and Pezizomycotina.To avoid word repetition,these taxonomic groups will be hereafter referred to as 13 taxon groups,and in the same order as shown in the list above.
Trimer usage probability scoring method
This method converts 3-codon DNA 9-mers in a query sequence into A.A.Char Trimers and identifies those with structural roles by searching them against Trimer Ref.DB.For each matching A.A.Char Trimer,the method first searches a matching A.A.Trimer,and second,a matching 3-codon DNA 9-mer in Trimer Ref.DB (Figure 3).It retrieves trimer usage biases of the matching 3-codon DNA 9-mers from CDS Genus-specific DB per reading frame of a query sequence.It repeats the same process with non-CDS Genusspecific DB.It then compares scores from CDS and non-CDS Genus-specific DBs,and selects a genus with the highest score (Figure 3).Users are provided with an option to include genera whose scores have little difference from the highest score.
Rank probability scoring method
This method measures a standardized 3-codon usage relative to an expected 3-codon usage computed from three individual mono codon usages.The Average A.A.Usage Table(20 amino acids and stop codons for 12 A.A.Char monomers) and the Average Codon Usage Table (64 codons for 20 amino acid monomers and stop codons)were created based on CDS database per genus.1296 Expected A.A.Trimer Usage Tables and 7674 Expected 3-codon Usage Tables were created based on the Average A.A.Usage Table and the Average Codon Usage Table,respectively (Figure 4).

Figure 2 Database design

Figure 3 Flow chart of the program
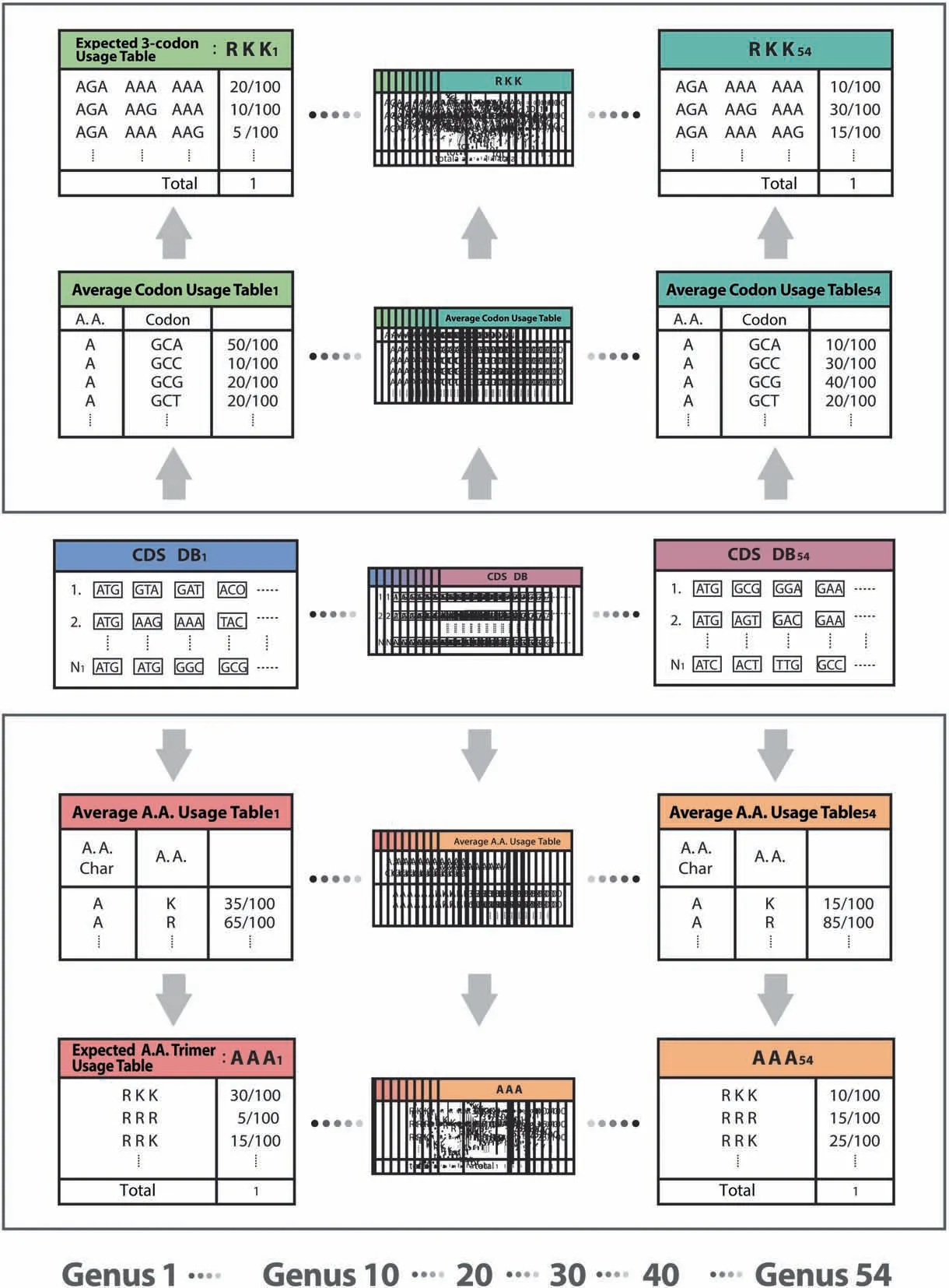
Figure 4 Creation of expected usage tables for the rank probability scoring method
A standardized 3-codon usage was calculated by dividing a 3-codon usage in a 3-codon Usage Table by an expected 3-codon usage in an Expected 3-codon Usage Table.Based on trimer usage biases and standardized 3-codon usages,we calculated a group mean for each taxon group and Kruskal Wallis(KW) test’s h-score on ranks of 13 taxon groups,from which we developed a rank probability score per 3-codon DNA 9-mer.In addition to the trimer usage biases,the rank probability scoring method adds another set of 224,383 scores per genus into Genus-specific DB.This method is applicable only to CDS.

Table 1 Conversion table from A.A.to A.A.Char
P value scoring method
We applied the concept of the sum of rolled numbers from a pair of dice to develop thePvalue scoring method (http://www.lucamoroni.it/the-dice-roll-sum-problem/).We drew analogies between the number of faces of a dice and 54 genera and between the number of dices we roll and the number of matching 3-codon DNA 9-mers identified in a reading frame of a query sequence.There were 54 possible ranks computed based on trimer usage biases per matching 3-codon DNA 9-mer.Pvalue scores were calculated based on a sum of ranks of matching 3-codon DNA 9-mers.Computational costs ofPvalues for all possible outcomes,sums of ranks,were too high.To reduce the computational costs,we approximatedPvalues.We obtained sample data per number of matching 3-codon DNA 9-mers based on Equation (1).

wherepis the sum of ranks,nis the number of dices per roll,sis the number of faces of the dice,54,and maximum ofkis](p-n)/s[ where ]x[ is the floor function (e.g.,]7.9[=7).We created a table ofPvalue scores per number of matching 3-codon DNA 9-mers.If a rank sum was less than one with the highestPvalue score,the approximate mean of all of the rank sums in each table,we multiplied thePvalue score with-1,indicating statistically non-significant outcome.In the test sets,the number of matching 3-codon DNA 9-mers varied widely,with a minimum of 30 and a maximum of 97.We have 624 tables in thePvalue score database covering 2-625 matching 3-codon DNA 9-mers.
Implementation and program availability
SeSaMe was implemented using the Java programming language (Java 8).We provided two sets of the programs.One requires Apache commons math3 (3.3) and IO (2.4) libraries(www.apache.org),while the other does not.The programs consist of executable Java JAR files and Java class files for Linux/Unix operating systems.SeSaMe has been tested andconfirmed to work on Linux system—CentOS Linux 7(www.centos.org)and is currently being used at the Biodiversity Center,Institut de Rechercheen Biologie Ve´ge´tale,De´partement de Sciences Biologiques,Universite´ de Montre´al.The trimer usage probability scoring method offered to the public produces output of smaller size,but is sufficient for the purpose of taxonomic classification and is freely available at www.fungalsesame.org.There are no restrictions to use the programs by academic or non-academic organizations as long as they comply with the terms and conditions of the license agreements.
Input,output,and options
SeSaMe utilizes a command-line interface.Input files should contain DNA sequence(s) in fasta format.The Java JAR files produce output files with sequence information (seq_id,matching A.A.Char Trimers,A.A.Trimers,and 3-codon DNA 9-mers) and genus information (rank,scores,andPvalue score).The output provides the information per reading frame per sequence.After processing the output file with Java class files,users are able to obtain a summary file containing one predicted outcome per query sequence.Java JAR files require users to give a mandatory command line argument— input file path.Java JAR files with the trimer probability scoring method may produce multiple genera as an answer if their scores have little difference.A user is given the option with 6 choices to select a cut-off value for the difference:0.01,0.05,0.1,0.15,0.2,or 0.3.Users can give the option to the Java class file called compare_result_coding_non_coding.class.The default cut-off value is 0.05.The lower the cut-off value is,the fewer genera will be included in an answer.
Results
We assessed the accuracy of the program by conducting classification experiments.We created metagenome test sets,ran the programs with them,and calculated the mean percentages of correct predictions.We showed the relationship between the correct prediction proportion and thePvalue score in order to provide users with useful examples in assessing the statistical significance of predicted outcomes.
Metagenome test sets
We randomly chose 100 sequences from each of the CDS and non-CDS databases per genus.We randomly selected a starting base pair position in each of the randomly chosen sequences.From the starting position,we randomly selected an ending base pair position so that a sequence length is within the range of 150-300 bp.Both of the CDS and non-CDS test sets consisted of 4500 bacterial and 900 fungal sequences (including AMF).
Mean percentages of correct predictions from the trimer usage probability scoring method
The mean percentages of correct predictions of the CDS and non-CDS test sets at the genus level were 71% and 50% for the bacterial group,68% and 73% for the fungal group (excluding AMF),and 49% and 72% for AMF,respectively.AMF had the lowest prediction percentage in the CDS genus test sets possibly due to a large number of heterogeneous nuclei and horizontal gene transfers from a variety of endobacteria during their evolution[8,10-14].The mean percentages of correct predictions at the genus level and at higher taxonomic ranks of the 13 taxon groups are shown inTable 2.
SeSaMe produced more than one genus as an answer per query sequence when multiple genera had little difference in their scores.We converted each predicted genus into one of the 13 taxon groups and calculated a proportion of the correct taxon group in answer per query sequence.We calculated the mean and the standard deviation of the proportions in each genus test set;1 represented that answers contained correct taxon groups only,while 0 represented that answers contained incorrect taxon groups only(Tables S3 and S4).The means of the bacterial and fungal CDS test sets were 0.87 and 0.57,respectively (Tables S3 and S4).
60% and 46% of the correctly predicted sequences contained only one genus as an answer in the bacterial CDS and non-CDS test sets,respectively (Figure S1;Table S5).90%and 76% of the correctly predicted sequences had a correct taxon group in the first rank in the bacterial CDS and non-CDS test sets,respectively (Figure S2;Table S6).Only 1%-5% of the sequences in the non-AMF test sets had AMF in an answer (Table S7).Although the trimer usage probability scoring method provides us not with the individual trimer usage biases but with result of multiplying all of the trimer usage biases,we can often derive general ideas about a query sequence from its answer(Figures S3 and S4).Does it contain only one genus in answer?Or what other genera does it contain in answer? For example,an AMF test sequence that containsClostridiumand AMF in answer may imply that the query sequence might have been acquired by horizontal gene transfer from a bacterium ancestor or thatClostridium’s ancestor might have become a heritable endosymbiont.
Mean percentages of correct predictions from the rank probability scoring method
The mean percentages of correct predictions of the CDS test sets were 82% for the bacterial group,72% for the fungal group (excluding AMF),and 42% for AMF.The mean percentages and the standard deviations of correct predictions of the CDS test sets were 64%±4.2%,71%±6.4%,84%±2.5%,70%±2.8%,73%±0%,83%±8%,74%±10%,81%±7.8%,88%±9.2%,85%±5.9%,42%±0%,65%±6.4%,and 79%±6.7% for the 13 taxon groups.Compared to the trimer usage probability scoring method,the rank probability scoring method produced the higher mean and the smaller standard deviation for the bacterial group.In general,the rank probability scoring method showed improvement in performance.Although the means for Clostridia and Gammaproteobacteria were lower,their standard deviations were much smaller in the rank probability scoring method:4.2%vs.22% and 7.8%vs.9.4%,respectively.The trimer usage probability scoring method showed better performance in Actinobacteria that had low withingroup variation of trimer usage bias.In contrast,the rank probability scoring method showed better performance in genera that had relatively flat peakness in a frequency distribution curve of synonymous 3-codon DNA 9-mers,in addition to genera that had relatively large within-group variation of trimer usage bias.
Relationship between correct prediction proportion and P value score
The means of the correct prediction proportions per number of matching 3-codon DNA 9-mers calculated based on the result of the trimer usage probability scoring method are shown in Figure S5A and Table S8.The means of correct prediction proportions per base 10 logarithm of an approximated inverse ofPvalue score (log10inverse ofPvalue score) calculated based on result of the trimer usage probability scoring method and of the rank probability scoring method are shown in Figure S5B,Table S9,Figure S5C,and Table S10,respectively.We divided the results of each genus test set into quartiles and calculated the range of log10inverse ofPvalue score and the mean and the standard deviation of the correct prediction proportions in each quartile (Tables S11 and S12).The first ranked genus with the highest probability score always had positivePvalue score.In general,as log10inverse ofPvalue score became higher —i.e.,as positivePvalue score became lower — the correct prediction proportion increased in all test sets.The frequencies of fungal sequences that had a correct taxon group in the 1st,2nd,3rd,4th,or 5th rank in an answer were comparable due to similarity of Dikarya(Figure S2;Table S6).Because the data for Figure S5B were generated based only on the first rank,the fungi showed relatively weak correlation between correct prediction proportion and log10inverse ofPvalue score.
Classification of an example sequence
The example sequence(AAATCCCAATGTCAGAATAAAG AAACTACCAGATGATCATCCTGTTTATCCTGGGTAT GGATTATTTGCTAACAAAGATCTTAAAAAATTTAAT CTAGTCGTTTGTTATACTGGCAAAGTTACAAAAAGA GAAATTGGGGGTGAAGAAGGAAGTGA) was selected from the AMF CDS test set and is 156 bp in length.The sequence had the highest trimer usage probability score in the second reading frame in the forward direction,which was then assumed as the open reading frame.SeSaMe identified 49 matching 3-codon DNA 9-mers that were matched to Trimer Ref DB.The program correctly classified the example sequence into CDS of AMF.Firmicutes,Cyanobacteria,Rickettsia,and AMF had higher trimer usage biases than Proteobacteria,Actinobacteria,and Dikarya in a majority of 3-codon DNA 9-mers.Figure 5shows trimer usage biases of the 11th 3-codon DNA 9-mer,GATGATCAT,in 54 genera.GATGATCAT belongs to A.A.Char Trimer,CCB and to A.A.Trimer,DDH.The multidimensional scaling (MDS)method was applied to a matrix containing trimer usage biases;it had 54 genera in rows and matching 3-codon DNA 9-mers identified in the open reading frame in columns (http://www.inf.uni-konstanz.de/algo/software/mdsj/) [34].It visualized proximity relationships among 54 genera in XY axis graph(www.jfree.org).It showed that Actinobacteria,Alphaproteobacteria,and Dikarya were compactly clustered,while Betaproteobacteria were spread out in the left side of the graph(Figure S6).Nostocales,Oscillatoriophycideae,Bacilli,and Clostridia were scattered across in the right side.AMF,Cyanobacterium,andRickettsiawere located in the far-right side.
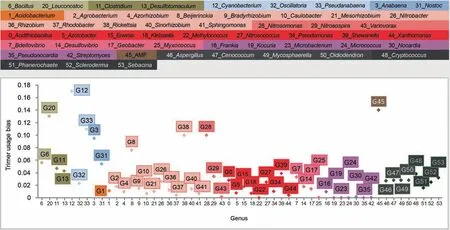
Figure 5 Trimer usage biases of the 11th 3-codon DNA 9-mer,GATGATCAT,in 54 genera
Future work
Microorganisms contain a number of heterogeneous alternative sigma factors that are selectively induced in response to environmental stress [35].They not only provide functionally specialized RNA polymerase subpopulations,but are also involved in regulating the expression of a set of target genes,or regulons [36,37].Ribosomal heterogeneity also has an important role in governing cellular stress.Sequence comparison of rRNA genes and ribosomal coding genes as well as sigma factor genes will be required in order to study their influence on adaptation of microorganisms [38,39].
Codon usage and codon context have been documented to play various important roles in microorganism’s adaptation to environment.If we sort the target genes in the CDS and in the non-CDS databases according to the alternative regulators,and create Genus-specific DB per group of the target genes,it may increase the accuracy of taxonomic classification.Moreover,comparative studies on alternative regulator subpopulations may provide useful insights into the development of genetic markers with which we can detect changes in microbial community structures in response to environmental stress(Figure S7).It may lead to new perspectives and strategies for improving the analysis of metagenome data,especially AMF inoculant field data sampled from highly stressful environments.
Data availability
SeSaMe is freely available at www.fungalsesame.org.
CRediT author statement
Jee Eun Kang:Conceptualization,Methodology,Software,Validation,Software,Writing-original draft.Antonio Ciampi:Supervision,Writing -review & editing.Mohamed Hijri:Supervision,Writing -review & editing.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
The authors gratefully acknowledge E´ ducation et de l’Enseignement supe´rieur Quebec (AFE),Faculte´ des e´tudes supe´rieures et postdoctorales de l’UdeM (FESP),and Institut de Recherche en Biologie Ve´ge´tale de l’Universite´ de Montre´al(IRBV) for awarding scholarships to KJE.The authors gratefully acknowledge insightful comments from Bachir Iffis,David Walsh,Etienne Yergeau,Franck Stefani,Ivan de la Providencia,Jesse Shapiro,Sylvie Hamel,and Yves Terrat.We also thank Andrew Blakney for English editing.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.07.010.
ORCID
0000-0003-2475-0474 (Jee Eun Kang)
0000-0003-4838-8297 (Antonio Ciampi)
0000-0001-6112-8372 (Mohamed Hijri)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- PIMD:An Integrative Approach for DrugRepositioning Using Multiple Characterization Fusion
- SeSaMe PS Function:Functional Analysis of the Whole Metagenome Sequencing Data of the Arbuscular Mycorrhizal Fungi
- iLBE for Computational Identification of Linear B-cell Epitopes by Integrating Sequence and Evolutionary Features
- GTB-PPI:Predict Protein-protein Interactions Based on L1-regularized Logistic Regression and Gradient Tree Boosting
- Exploring Potential Signals of Selection forDisordered Residues in Prokaryotic and Eukaryotic Proteins
- Pooled Plasmid Sequencing Reveals the Relationship Between Mobile Genetic Elements and Antimicrobial Resistance Genes in Clinically Isolated Klebsiella pneumoniae