高速逆流色谱法分离制备茯苓皮中茯苓酸A 和茯苓酸B
2020-08-26殷梦舟张莉佳
殷梦舟,刘 莹,张莉佳,王 益,黄 文*
(华中农业大学食品科学技术学院,湖北 武汉 430070)
茯苓皮为多孔菌科真菌茯苓(Poria cocos(Schw.)Wolf)干燥的菌核外皮,2015版《中国药典》中记载其味甘、淡、平,归肺、脾、肾经,有利水消肿的功效[1],是一种常见的利尿中药。茯苓皮作为茯苓加工过程的副产物,其利用率很低,甚至被作为下脚料废弃。研究表明,茯苓皮与茯苓菌核中的成分存在显著差异,茯苓多糖主要存在于茯苓菌核中,而三萜酸类化合物主要存在于茯苓皮中[2-4]。目前从茯苓皮中已经分离出超过40 种三萜酸[5-6]。近年来的药理研究表明,茯苓三萜酸类化合物药理作用广泛,具有抗肿瘤[7-9]、抗炎[10-11]、保肝[12]、利尿[13-14]等生理活性。
天然产物是新药先导化合物发现的重要资源之一[15],而制备色谱技术是从天然产物中获得大量高纯度单体必不可少的方法,是对化合物进行结构信息和生化性质研究的基础。经过100多年的发展,分离色谱技术已经从薄层色谱、常压柱色谱逐步发展到高压柱色谱、逆流色谱等,成为了科学研究和生产实践中分离复杂组分的一种重要技术手段[16-17]。以茯苓皮三萜为例,三萜类化合物在茯苓皮中种类众多,结构相近,分离纯化较为困难。胥秀英等[18]采用两次硅胶柱层析,制备高效液相色谱(high performance liquid chromatography,HPLC)纯化的方法从茯苓皮中分离鉴定得到乙酰茯苓酸和猪苓酸C。杨丹等[19]使用硅胶柱色谱分两步从茯苓皮的正丁醇萃取部位分离鉴定出9 种三萜类化合物。上述方法虽然可以实现茯苓皮中三萜类化合物的分离,但存在过程繁琐,得率不高,溶剂消耗量大的缺点。
逆流色谱是由Ito等[20]在20世纪70年代早期发明一种基于液-液分配的色谱方法,它为分离纯化方法提供了一种新的方法模式。与传统以固体为载体的分离方法相比,逆流色谱克服了固体载体不可逆吸附的缺点,因此,样品回收率得到很大提升。除此之外,逆流色谱还具有溶剂消耗少,对样品颗粒度接受能力强等优点[21]。高速逆流色谱是由逆流色谱发展而来,与逆流色谱相比,其在分辨率、分离时间和样品加载能力上都有了很大提升,现在已经被广泛应用于天然产物的分离纯化。本实验利用高速逆流色谱技术,根据目标化合物筛选所需溶剂系统,并对分离过程的流速、上样量参数条件进行优化以制备茯苓皮三萜酸类物质,进一步采用液相色谱、质谱以及核磁共振对三萜类化合物进行鉴定,研究结果将为茯苓皮中三萜酸的高效制备提供基础。
1 材料与方法
1.1 材料与试剂
茯苓皮采自湖北咸宁;正丁醇、正己烷、乙酸乙酯、甲醇、磷酸(均为分析纯) 国药集团化学试剂有限公司;甲醇(色谱纯) 赛默飞世尔科技有限公司;水为二次蒸馏水。
1.2 仪器与设备
RE2000A旋转蒸发器 上海亚荣生化仪器厂;HPLC仪、超高液相色谱-离子淌度四极杆飞行时间质谱联用仪、e2695二极管阵列检测器 美国Waters公司;TBE-300C型高速逆流色谱仪 中国上海同田生化技术有限公司;AVANCE III HD 600 MHz核磁共振仪 美国Bruker公司。
1.3 方法
1.3.1 茯苓皮乙酸乙酯萃取物的制备
茯苓皮洗净烘干,用中药粉碎机粉碎,称取一定量茯苓皮粉于提取瓶中,按液料比30∶1(mL/g)加入正丁醇-水(3∶1,V/V)溶液于60 ℃回流提取1 h,过滤除去滤渣,分离滤液取正丁醇相,旋转蒸发得到红棕色浸膏。浸膏用蒸馏水悬浮,加入乙酸乙酯萃取3 次,合并乙酸乙酯相,旋转蒸发浓缩,低温烘干得到乙酸乙酯萃取物,4 ℃避光保存备用。
1.3.2 HPLC测定条件
取一定量茯苓皮乙酸乙酯萃取物和高速逆流色谱分离样品溶于色谱甲醇中,经0.45 μm针头过滤器过滤后进行HPLC分析,采用面积归一法确定化合物的纯度。
HPLC检测条件:InertSustainTMAQ-C18色谱柱(4.6 mm×250 mm,5 μm);流动相A为水(含0.05%磷酸),流动相B为甲醇;梯度洗脱:0~15 min,30%~25% A、70%~75% B;15~25 min,25%~21% A、75%~79% B;25~40 min,21%~10% A、79%~90% B;40~65 min,10% A、90% B。流速1 mL/min,柱温25 ℃,检测波长242 nm,进样量10 μL;紫外光谱扫描范围210~400 nm。
1.3.3 高速逆流色谱溶剂系统的选择
1.3.3.1 溶剂系统的确定
根据目标化合物的性质,参考高速逆流色谱分离三萜类化合物溶剂系统的相关报道[22-23],选择不同比例的正己烷-乙酸乙酯-甲醇-水溶剂系统进行实验,考察溶剂系统的分层时间、对目标物质的分配系数确定用于高速逆流色谱的溶剂系统。
1.3.3.2 溶剂系统上下相分层时间的测定
室温下分别取上下相各5 mL于具塞试管中,将其倒转5 次,使上下两相充分混合后垂直置于桌面静置分层,测定其两相界面清晰的时间,重复测定3 次取平均值。
1.3.3.3 溶剂系统分配系数K的测定
取等体积充分平衡的溶剂系统上下相,加入少量茯苓皮乙酸乙酯萃取物,充分振荡使样品完全溶解在溶剂系统中;平衡后分离两相,分别减压旋转蒸发蒸干溶剂,溶解在甲醇中通过HPLC分析。分配系数K值定义为目标化合物在上相中的色谱峰面积(S上)与目标化合物在下相中的色谱峰面积(S下)比,即K=S上/S下。
1.3.4 高速逆流色谱分离条件的确定
在高速逆流色谱分离过程中,温度、主机转速、进样量、流速等分离条件对化合物的分离效率均有影响。在预实验的基础上,保持高速逆流色谱主机正转,转速800 r/min,根据在波长242 nm处采集分离图谱进一步考察流速(2、3、4 mL/min)和进样量(50、100、150 mg)对分离效率的影响,确定分离条件。
1.3.5 高速逆流色谱法分离制备茯苓皮中三萜类化合物
按一定比例配制正己烷-乙酸乙酯-甲醇-水组成的两相溶剂系统,在分液漏斗中充分振摇混合后静置,分离出上下两相,上相作为固定相,下相作为流动相,两相超声脱气15 min,冷却至室温待用。以30 mL/min的流速将固定相泵入主机中,待出口流出20~50 mL液体后,启动主机正转,调整转速为800 r/min,将检测器波长设置为242 nm,同时开始以流速4 mL/min泵入流动相,待流动相流出主机,记录流出的固定相体积,保留率按下式计算:

取一定量茯苓皮乙酸乙酯萃取物溶解于10 mL下相中,设定分离流速,待基线稳定后由进样阀进样,打开色谱工作站采集数据,收集各分离组分。
1.3.6 化合物鉴定
通过超高效液相色谱-离子淌度四极杆飞行时间质谱、核磁共振氢谱(1H nuclear magnetic resonance,1H NMR)和核磁共振碳谱(13C nuclear magnetic resonance,13C NMR)对高速逆流色谱分离得到的化合物进行鉴定。
超高效液相色谱条件:ACQUITY UPLC®BEH C18色谱柱(2.1 mm×100 mm,1.7 μm);流动相A为水,流动相B为甲醇;梯度洗脱:0~7 min,44%~23% A、5 6%~7 7% B;7~1 4 m i n,2 3%~1 5% A、77%~85% B;14~24 min,15% A、85% B。柱温30 ℃,进样量1 μL,流速0.3 mL/min。
质谱条件:负离子模式,干燥气温度350 ℃,干燥气流量600 L/h,毛细管电压2.4 kV,质量扫描范围m/z10~1 000,根据一级质谱和二级质谱得到化合物母离子和其他碎片离子的信息[24]。
核磁共振的样品溶剂为氘代甲醇,谱图数据采用Meatrenove 14.0进行分析处理。通过上述数据与相关报道进行比对,确定化合物的结构,化合物的结构式通过ChemDraw 16.0绘制。
2 结果与分析
2.1 HPLC分析

图1 茯苓皮乙酸乙酯萃取物的HHPPLLCC图Fig. 1 HPLC chromatogram of the EtOAc-soluble fraction of Poriae Cutis
由图1可以看出,茯苓皮乙酸乙酯萃取物的化学成分比较复杂,为了方便后续溶剂系统的优化,选择峰面积较大的5 个峰作为分离纯化的目标化合物,其保留时间分别为33.46、34.76、36.34、52.73、56.51 min。
2.2 高速逆流色谱溶剂系统的选择
高速逆流色谱的应用中溶剂系统的选择对于样品中不同组分的分离具有决定性的作用,不同的溶剂系统由于极性、密度、黏度的不同都会对化合物的溶解性、分配能力产生影响。所选的溶剂系统应满足以下要求[25]:样品可以稳定的溶解在溶剂系统中;溶剂体系能形成稳定的上下两相,且体积比合适;溶剂系统能对目标化合物提供合适的分配系数;固定相在两相平衡中有较高的保留率。考虑到目标化合物的极性,按照1.3.3节方法分别测定3 种溶剂系统两相分层时间以及对峰1~5五种化合物的分配系数,结果见表1。
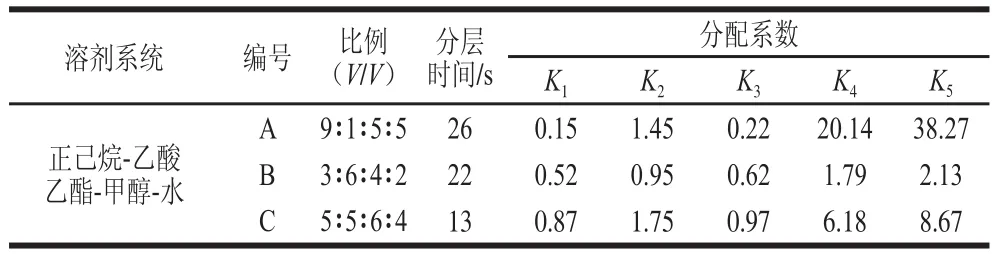
表1 茯苓皮提取物在不同溶剂体系中的分配系数Table 1 Partition coeffificients ( ) of the extract of Poriae Cutis in different two-phase solvent systems
在高速逆流色谱分离中,要获得好的分离效果和较高的分离效率,一般要求溶剂系统的分层时间较短(小于30 s),固定相保留率在40%以上,溶剂系统对目标化合物的分配系数在0.5~2之间比较合适[26]。由表1可知,3 种溶剂系统的分层时间都少于30 s。溶剂系统A对化合物1和3的分配系数小,而对化合物4和5的分配系数又过大,表明这些目标化合物在溶剂系统中在某一相中溶解性较好,这不利于这些化合物的分离。C溶剂系统对化合物4和5的分配系数较大,使其较好地溶解在固定相中,分离时间长,不利于其分离。因此综合考虑以上因素,选择溶剂系统B作为本实验的高速逆流色谱溶剂系统。
2.3 高速逆流色谱操作条件的选择
在高速逆流色谱分离过程中,除溶剂系统外,分离条件也直接影响分离结果。在高速逆流色谱主机正转,转速800 r/min时,分别在不同流速(2、3、4 mL/min)和不同进样量(50、100、150 mg)条件下采集分离过程中波长242 nm处的色谱图。比较分离结果发现,增加分离过程的流速可以缩短分离所需时间,当流速达到4 mL/min时,峰III和峰IV的分离度较低,分离过程中可能会相互影响使分离得到的化合物纯度不高,因此分离流速选择3 mL/min。除此之外,进样量的增加使得各分离峰的峰面积增大,当进样量达到150 mg时,出峰时间接近的峰III和峰IV相互重合,无法分离。综上所述,本实验采用高速逆流色谱分离操作条件进样量100 mg、流速3 mL/min,分离图谱如图2所示。

图2 茯苓皮乙酸乙酯萃取物高速逆流色谱图Fig. 2 HSCCC chromatograms of the EtOAc-soluble fraction of Poriae Cutis
2.4 高速逆流色谱分离结果

图3 峰III(a)、峰IV(b)组分的HHPPLLCC图Fig. 3 HPLC chromatograms of eluate fractions III (a) and IV (b)
采用正己烷-乙酸乙酯-甲醇-水(3∶6∶4∶2,V/V)的溶剂系统,按照优化的分离操作条件进行高速逆流色谱分离,平衡过程共流出固定相150 mL,该型仪器的总柱体积为300 mL,按1.3.5节方法计算保固定相保留率为50%。将峰I~IV对应的组分进行HPLC分析,结果表明峰I和峰II的杂质较多,三萜含量较少,没有进一步分析价值。峰III和峰IV三萜的纯度较高,其保留时间和纯度分别为:峰III,33.23 min,90%;峰IV,36.20 min,92%(图3)。由对应插图可知,2 个色谱峰均在波长242 nm处有最大吸收,符合三萜类化合物的紫外吸收特征。
2.5 结构表征
将高速逆流色谱分离得到的2 个组分进行冷冻干燥后,得1号化合物5.2 mg(峰III)和2号化合物20.5 mg(峰IV)。经超高液相色谱-离子淌度四极杆飞行时间质谱、1H NMR和13C NMR鉴定1号化合物为茯苓酸B,2号化合物为茯苓酸A,如表2、3和图4所示。

表2 化合物1的H NMR和13C NMR化学位移Table 2 H NMR and 13C NMR chemical shifts of compound 1

表3 化合物2的H NMR和13C NMR化学位移Table 3 H NMR and 13C NMR chemical shifts of compound 2
化合物1:白色无定型粉末(甲醇)。化合物在紫外光谱中的最大吸收波长为242 nm,ESI-MSm/z:483 [M-H]-,409 [M-CH3CH2COOH]-。如表2所示,1H NMR谱中δH 1.50、1.58、0.99、0.93、1.50、1.68为6 个甲基单峰质子,δH 4.67、4.76为环外双键质子,δH 5.17、5.21为环内双键质子,推测该化合物具有共轭双键结构,δH 4.54为与羟基相连的碳原子上的质子信号。13C NMR谱中共显示30 个碳信号,δC 178.3、180.0为2 个羧基所在位置的碳信号,δC 119.0、120.7和142.3、138.3分别为2 个烯属的次甲基和2 个烯属季碳的共振信号,结合1H NMR谱信息推测该化合物为3,4-开环-羊毛甾-7,9(11)-二烯型三萜。综合上述分析,与相关文献[27-28]对照,鉴定化合物1为茯苓酸B。

图4 茯苓酸A和茯苓酸B的化学结构Fig. 4 Chemical structure of poricoic acid A and poricoic acid B
化合物2:白色无定型粉末(甲醇)。化合物在紫外光谱中的最大吸收波长为242 nm,ESI-MS m/z:497 [M-H]-,423 [M-CH3CH2COOH]-。如表3所示,其数据与化合物1极为相似,推测化合物2与化合物1具有相同的骨架类型。13C NMR谱中数据与化合物1相比,δC 25 132.6向高场移至33.6,δC 24 124.9向低场移至155.3,同时由化合物2中δC 31 106.7推测其环外的双键类型由化合物1中的24(25)型双键变成24(31)型的双键。以上数据与相关文献报道[29-30]对照基本一致,故鉴定化合物2为茯苓酸A。
3 结 论
茯苓皮作为茯苓加工的副产物,其中的活性成分三萜类化合物的总含量达到1.58%[31],因此开发利用茯苓皮中的三萜类化合物具有重要意义。茯苓三萜分离制备的传统方法主要依靠柱色谱、薄层色谱等方法相结合进行反复的分离纯化,鉴于这些方法存在步骤复杂,溶剂消耗量大,得率较低的缺点,有研究者采用高速逆流色谱技术分离茯苓中的三萜类化合物。王艳等[32]以大孔树脂初分后茯苓皮乙醇提取物为样品,利用高速逆流色谱在5 h内分离得到了纯度为98%的茯苓酸A;Dong Hongjing等[33]采用酸碱萃取的方法将茯苓醇提物分为两部分进行高速逆流色谱分离,分别在5 h和7 h内共分离得到纯度为80.1%到96.4%的6 种三萜酸组分。综上,有关于高速逆流色谱分离茯苓皮中三萜类化合物的报道较少,尚缺少溶剂系统筛选、分离条件优化以提高分离效率的报道。
本实验应用高速逆流色谱从茯苓皮中分离出了2 种三萜类化合物茯苓酸A和茯苓酸B。高速逆流色谱的溶剂系统为正己烷-乙酸乙酯-甲醇-水(3∶6∶4∶2,V/V),分离条件为主机正转、转速800 r/min、进样量100 mg、流速3 mL/min,在3 h内即完成分离。经质谱和核磁鉴定2 种组分分别为茯苓酸A和茯苓酸B,其纯度分别达到92%和90%。本实验建立了一种快速有效分离制备茯苓皮中茯苓酸A和茯苓酸B的高速逆流色谱方法,为进一步研究茯苓中的化学成分、药理活性及其有效成分的加工利用提供一定的理论基础。
