基于近红外法的紫胶原胶主组分快速预测模型构建
2020-08-24唐保山张雯雯关庆芳史正军
唐保山,李 坤, 张 弘, 张雯雯, 关庆芳, 史正军
(1.西南林业大学 林学院,云南 昆明 650224; 2.中国林业科学研究院资源昆虫研究所国家林业和草原局特色森林资源工程技术研究中心,云南 昆明 650233;3.安宁戴科精细化工有限公司,云南 昆明 650301)
紫胶又名虫胶、赤胶、紫草茸,是紫胶虫吸食寄主植物汁液后分泌的一种纯天然树脂混合物[1-4]。紫胶是我国重要的工业原料,因其具有优良的抗酸、防锈、耐油、电绝缘性和热塑性能,广泛用于国防、涂料、机械、医药、印刷和食品行业[5-7]。依据紫胶的不同加工阶段和产品形态,紫胶又可分为紫胶原胶、颗粒紫胶、片胶、漂白紫胶等。紫胶原胶是直接从寄主树上采收下来的成熟胶梗,也是紫胶其他产品加工的原料,主要由紫胶树脂、蜡质和色素组成[8-10]。长期以来,紫胶原胶的质量评价多依据国标法,尽管国标法具有可靠性、普适性强的特点,但操作复杂、对人员和熟练程度要求高、检测耗时等缺点已在很大程度上妨碍了国标法在行业,尤其在流通领域及企业中的普及和应用。在工业快速发展的今天,如何提高紫胶检测方法的准确性、时效性和便携性已成为提高工作效率的重要环节[11-12]。近红外光谱法是近年发展起来的一种非破坏性分析技术,具有检测成本低、速度快、样品预处理简单等优点[13-15]。近红外光谱分析法是一种间接分析技术,是用统计学方法在样品待测属性值与近红外光谱数据之间建立一个关联模型,之后通过验证完成模型的建立,利用近红外光谱仪测得的样品光谱数据即可通过软件模型预测被测组分含量[16-17]。目前,近红外光谱法已在药品、纺织物、酒类、饲料、粮油、烟草等领域得到广泛的应用[18-19]。其中饲料中水分、粗蛋白质、粗纤维等含量的测定[20],玉米中淀粉含量的测定[21]已成为国标中采用的标准检测方法。本研究依据化学法测定紫胶原胶中树脂、蜡质和色素含量的真实值,探索基于近红外光谱分析模型的构建方法,以期将近红外光谱法用于紫胶的检测,实现快速定量检测紫胶原胶中主组分含量的目的,为建立科学化、自动化的紫胶现代质量标准评价体系提供理论依据。
1 实 验
1.1 原料、试剂与仪器
紫胶原胶样本(中国21、泰国2、印度2、缅甸2和越南2),由墨江县洪森虫胶有限公司、墨江滇力林化厂、墨江森源科技有限公司、绿春兴龙紫胶有限公司、云县凝鑫紫胶产业发展有限公司和安宁戴科精细化工有限公司提供。为保证测试样品的均匀性,把收集到的29个紫胶原胶样本分别用高速粉碎机粉碎,取粒径≤0.38 mm的样品备用。无水碳酸钠、盐酸、邻苯二甲酸氢钾、四氯化碳、95%乙醇、无水乙醇和氢氧化钾均为分析纯。
MPA傅里叶变换近红外光谱仪,德国Brucker公司;Agilent Cary Seties紫外(UV-vis)可见分光光度计,Agilent Technologies公司;HH- 601水浴磁力搅拌器;Testo-206便携式pH计;SHZ-D(Ⅲ)循环水式多用真空泵。
1.2 紫胶中主组分的测定
1.2.1紫胶树脂 紫胶树脂的测定参照国标GB/T8143—2008[22]的方法进行。精确称取样品5 g放入用95%乙醇萃取过的定性滤纸中,用棉线捆紧后放入事先在100 ℃质量恒定的萃取瓶中,置入索氏提取器,用95%乙醇在80 ℃提取4 h,将萃取瓶中的溶剂减压蒸发,在100 ℃下干燥至质量恒定,最后扣除蜡质质量,即得紫胶树脂质量,平行测定3次。树脂质量分数按式(1)计算。
1.2.2紫胶蜡 紫胶蜡的测定参照国标GB/T8143—2008[22]中的方法进行。精确称取样品10 g放入烧杯中,之后加入150 mL溶有2.5 g碳酸钠的热水,放在沸水浴中加热搅拌。待试样溶解后再加热2~3 h(不要搅拌),然后将烧杯从水浴中取出,冷却至室温,蜡质结晶析出,用四氯化碳萃取过的滤纸过滤,用水洗涤至滤液无色,将滤纸取出放在60 ℃的烘箱中烘去水分,再用一张滤纸包好,用棉线捆紧,放入事先在100 ℃质量恒定的萃取瓶中,置入索氏提取器,用四氯化碳在80 ℃提取4 h,将萃取瓶中的四氯化碳减压蒸发,放入干燥箱中,100 ℃下干燥至质量恒定,平行测定3次。蜡质量分数按式(2)计算。
ω1=(m2-m1-m0)/m×100%
(1)
ω2=(m3-m1)/m×100%
(2)
式中:ω1—紫胶中树脂质量分数,%;m—样品质量,g;m0—蜡质质量,g;m1—萃取瓶质量,g;m2—萃取瓶与树脂质量和,g;ω2—紫胶中蜡的质量分数,%;m3—萃取瓶和蜡质质量和,g。
1.2.3紫胶色素 参照文献[23]测定水溶物。精确称取样品10 g放入250 mL烧杯中,加入150 mL去离子水浸泡过夜,之后抽滤,用水洗至无色,最后滤液倒入烧杯中在80 ℃鼓风干燥箱烘至质量恒定,平行测定3次。水溶物质量=烧杯与水溶物质量之和-烧杯质量。
参照国标GB1886.17—2015[24]方法进行色价测定。称取干燥水溶物0.1 g置于50 mL烧杯中,加入10 g/L碳酸钠溶液10 mL搅匀、溶解,倾入100 mL容量瓶中,用少量水洗涤烧杯,洗涤液并入100 mL容量瓶中,用水稀释至刻度、摇匀。准确吸取5 mL置于100 mL容量瓶中,用0.1 mol/L盐酸溶液调pH值至3.0左右,用pH值3.0缓冲溶液稀释至刻度、摇匀,即为试样溶液。取出试样溶液置于1 cm比色皿中,于紫外分光光度计为490 nm波长处测量吸光度,平行测定3次。色价按式(3)计算。
参照文献[25]方法测定色素含量。已知色素标准品的含量,测出色素标准品的色价,样品通过测定其色价,按照色素标准品折合试样色素含量,最后计算出紫胶原胶中的色素,平行测定3次。色素按式(4)计算。
(3)
ω3=m4/m×ω4
(4)

1.3 模型的建立

1.3.2模型建立的方法和步骤 一个完整的近红外预测模型包括6个部分,具体包括:样本数据采集及样品集划分、近红外光谱测定条件设定及光谱图采集、确定数据样本预处理方法、样品近红外光谱区域的划分及最优波段的选取、主成分数的选择和模型质量评价。
1.3.2.1样本数据采集及样品集划分 所收集的样本见表1,囊括了中国、越南、泰国、缅甸和印度等紫胶主产国,为基于近红外法的紫胶原胶主组分快速预测模型构建提供了数据支撑。
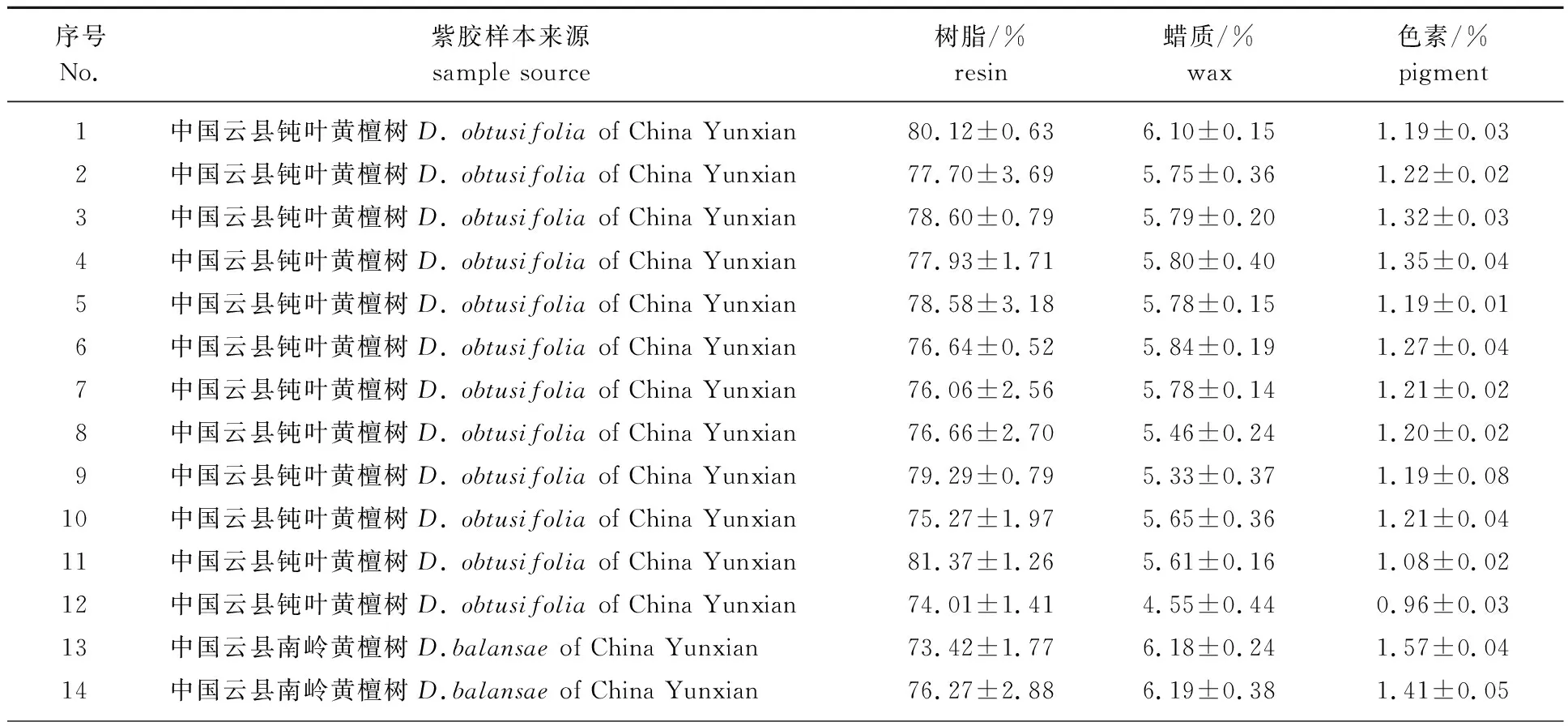
表1 不同来源紫胶样本中树脂、蜡质及色素含量1)
紫胶样本按4.5 ∶1随机分选为校正集和验证集,通过表2可知,校正集和验证集中树脂、蜡质和色素的含量范围、平均值和极差表明,所选样品中各组分含量范围较宽,代表性较强,且校正集样品范围包含验证集所有样品的组成范围,符合建立模型样品选择的原则。

表2 紫胶原胶中树脂、蜡质和色素样品集的划分
1.3.2.2测定条件设定及光谱图采集 为保证仪器的稳定性,开机后预热30 min;设置扫描时仪器分辨率为8 cm-1,扫描次数为64次,光谱范围为波数12500~4000 cm-1;以空IN312-SH型样品杯(Φ=97 mm)作为参比样品进行光谱采集,上样量以样品体积的2/3为宜,为确保样品采集的均匀性,采用旋转模式,每个紫胶原胶样品重复采集3次[27],取其平均光谱作为样品代表性光谱。
1.3.2.3确定数据样本预处理方法 近红外光谱易受到样品的均匀性、噪音信号以及杂散光等影响,为保证谱图基线平稳和所建立校正模型的准确性,必须对原始光谱进行预处理。常用的预处理方法有无光谱处理、多元散射校正(MSC) 、消除常量偏移量(ECO)、矢量归一化(VN)、最小-最大归一化(M-MN)、一阶导数+矢量归一化 (FD+VN)[28]。
1.3.2.4区域划分及最优波段选取 偏最小二乘法(PLS) 虽然可以处理全谱信息,但全谱中往往包含了过多的冗余信息,这既影响了建模的准确性,过大的数据量也降低了模型预测的计算速率[29]。因此,需要对光谱波段进行提取,改善模型性能。依据紫胶原胶主组分近红外谱图的吸收特征,将原始光谱分为5个波段范围进行交互建模,并考察各个波段的模型与组分含量间的相关性,探索最优波段,5个 波段分别为9400~7500、7500~6100、6100~5450、5450~4600和4600~4250 cm-1。
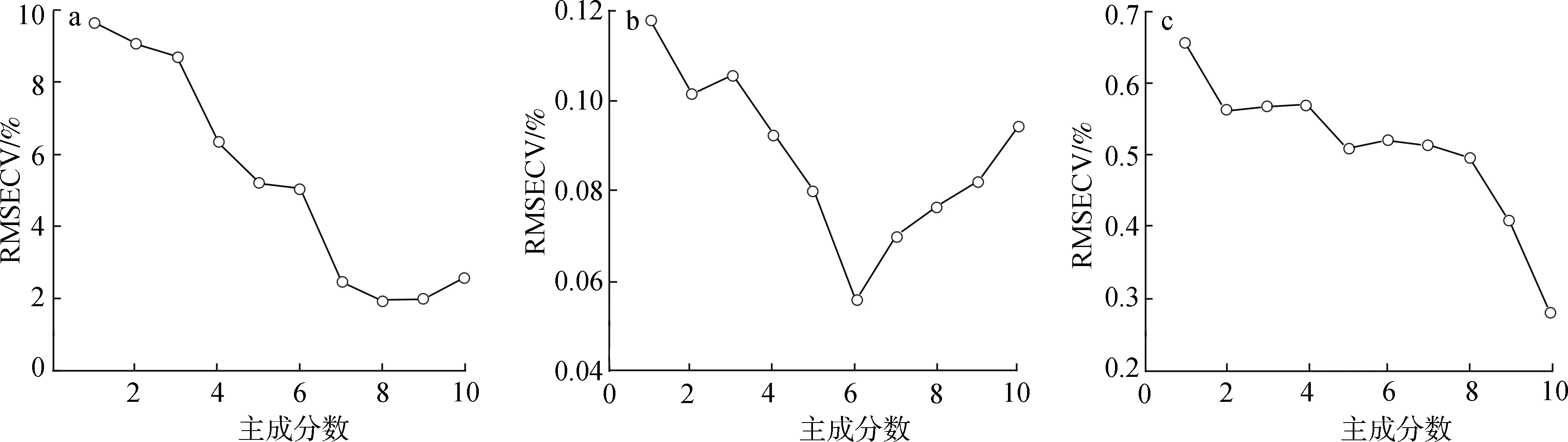
1.3.2.5主成分数的选择 在建模过程中,采用不同的主成分数,模型的预测能力也会有较大的差异。因此合理确定参加建模的主成分数是充分利用光谱信息和滤除噪音的有效方法之一。通常数值过大,所建模型包含太多的噪音,会出现过拟合现象;主成分太小,则导致建模信息不全,模型预测能力相对较差。当RMSECV最小时,对应主成分数最佳。

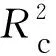
(5)
(6)
(7)
(8)
2 结果与分析
2.1 光谱的预处理
利用近红外仪器自带的OPUS 7.5分析软件采集的紫胶样品的近红外光谱,原始光谱如图1(a)所示。从图1(a)可以看出,紫胶主组分在峰位、峰强和峰形等方面重叠严重,很难直观分析,就需要对近红外光谱信号进行分析处理,不同的预处理方法得到的谱图见图1(b)、1(c)和1(d)所示。

通过对不同波段的筛选,由表3可知,树脂、蜡质和色素的最优建模波段分别为9403.7~5446.3、4601.6~4246.7 cm-1,9403.7~7498.3、6102~5446.7 cm-1和7502.1~6098.1、5450.1~4246.7cm-1,说明在该波数范围内,组分与模型的相关性最佳。
2.2 各个模型的参数及测定效果
为更好的验证上述最优波段选择的可靠性,选择了脱蜡脱色片胶、紫胶蜡和紫胶红色素作为紫胶树脂、蜡质和色素的纯净物分别进行全谱扫描。

a.紫胶原始图original spectra of sticklac; b.矢量归一化处理vector normalization (VN);c.最小-最大归一化处理
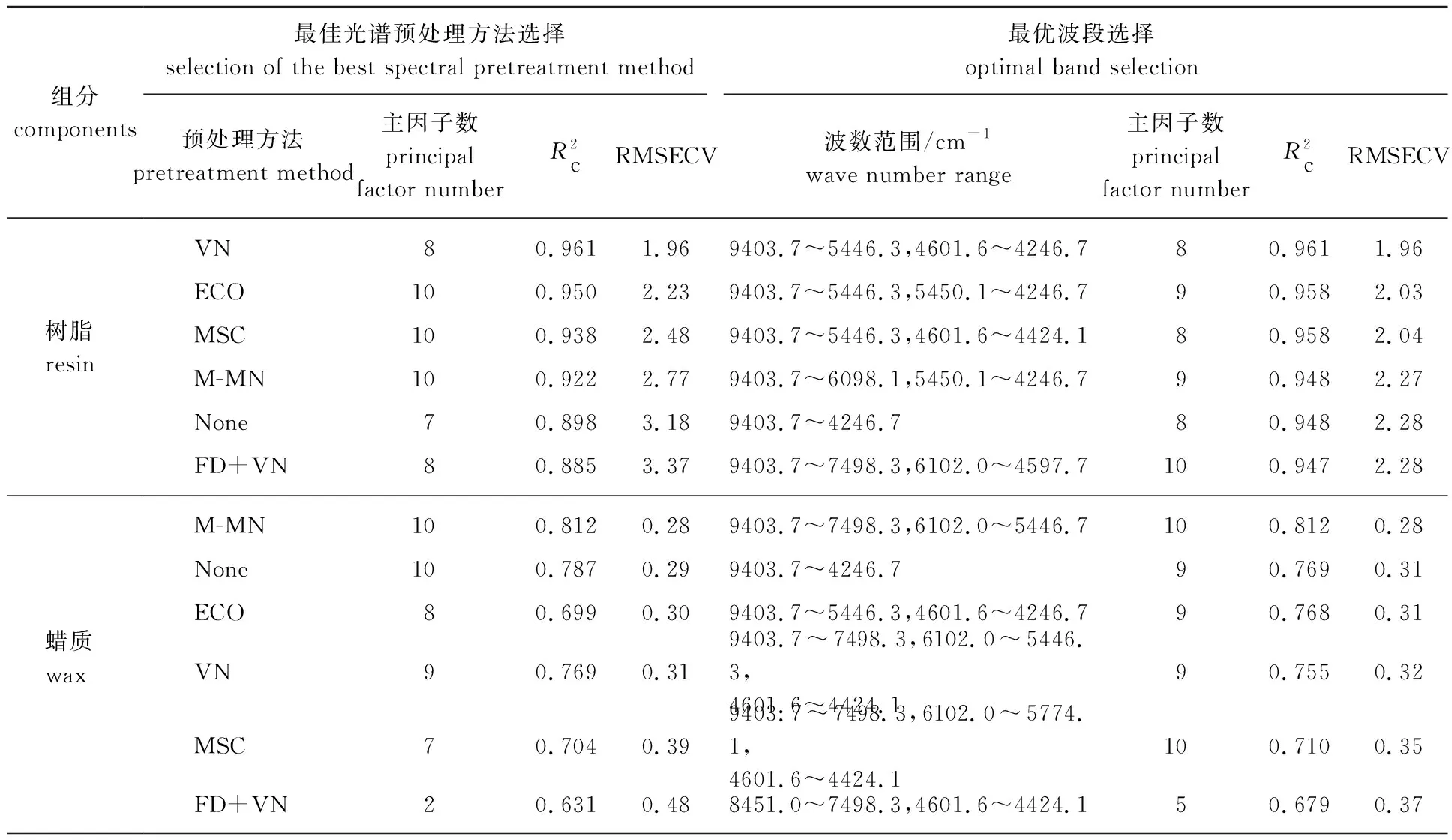
表3 树脂、蜡质和色素最佳预处理方法和最优波段的选择

续表3

a.树脂resin; b.蜡质wax; c.色素pigment图2 不同样品的近红外光谱图Fig.2 Near infrared spectra of different samples
由图2可知,纯树脂的近红外光谱图与紫胶原胶的光谱图几乎一致,是因为紫胶原胶中树脂是主要组分,树脂吸收强度最强,在谱图叠加中占据主导,故从纯树脂的光谱图上很难找到最优波段。由表3得到蜡质的最优建模波数为9403.7~7498.3 cm-1、6102.0~5446.7 cm-1,对比图2蜡质图谱可知,此波数范围正好在蜡质特征吸收范围内。由色素近红外光谱可知,色素主要吸收波段为7378.0~6258.0 cm-1和5411.0~4511.0 cm-1,而通过不同光谱段处理得到最优波段为7502.1~6098.1、5450.1~4246.7 cm-1,与色素主要吸收波段吻合。
树脂、蜡质和色素的主成分数与RMSECV的关系如图3所示,主成分数的大小影响模型的质量,一般认为RMSECV最小时,对应主成分数最优。由图3可知,树脂、蜡质和色素主成分数分别为8、10、6时,对应的RMSECV最小,说明在该主成分数下,该模型的误差最低。
a.树脂resin; b.蜡质wax; c.色素pigment
2.3 模型验证
利用以上建模条件,对树脂、蜡质和色素校正集进行内部交叉验证,建立校正模型的样品数为24个,分别剔除0、0、2个光谱异常的样品,实际采用的样品只有24、24、22个,利用建立的树脂、蜡质和色素定量模型预测验证集为5个样品。树脂、蜡质和色素的校正集和验证集的预测值和真实值的关系如图4所示。

△校正集calibration sets; ○验证集validation sets
所建模型的各项数理统计指标都达到预期值,所有样品的近红外光谱测定结果和标准方法之间的相关性以及误差基本都在要求的再现性范围以内,近红外光谱所建紫胶原胶中3种主组分模型预测样品的预测值与国标方法测得的化学值基本一致,说明所建模型的预测效果良好。
由表4可知,对树脂、蜡质和色素的预测值和真实值做了比较,每个样品的5次预测结果平均差异都小于0.10%;并且对每个样品的5次预测值做了显著性分析,结果P>0.05,均无显著性差异,可见所建模型精密度和稳定性良好。
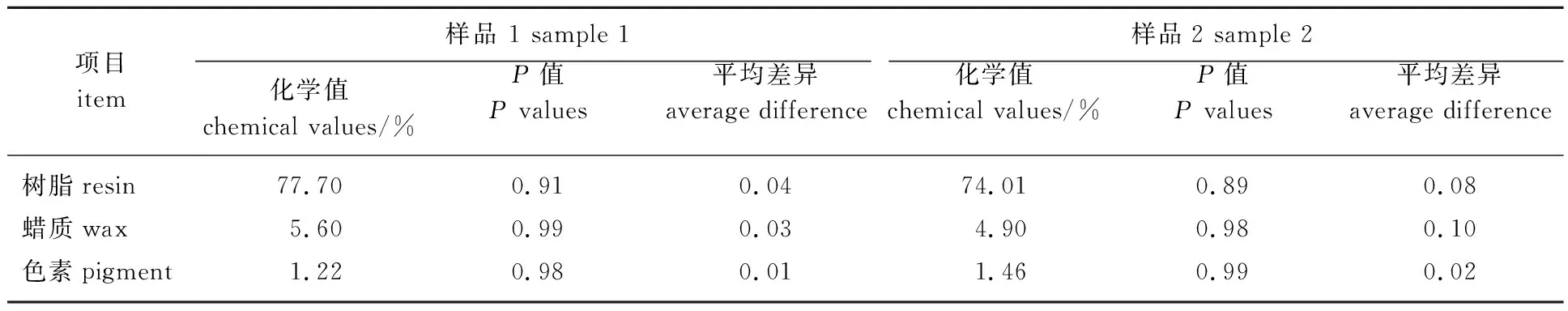
表4 模型预测样品的精密度和稳定性检验(n=5)
3 结 论

