次黄嘌呤和别嘌醇的太赫兹光谱弱相互作用研究
2020-08-21张琴陈涛
张琴 陈涛
摘 要 应用太赫兹时域光谱(THz-TDS)技术测量了次黄嘌呤(HPX)和别嘌醇(ALP)两种同分异构体在室温条件下0.1~2.0 THz范围的THz吸收光谱。同时,为进一步分析样品在THz波段的低频振动模式和弱相互作用类型,借助密度泛函理论(DFT)对两者结构进行几何优化,应用势能分布(PED)分析对分子基团的振动模式进行归属,并应用基于分子力场的能量分解分析(EDA-FF)方法对光谱色散特性进行定性分析。PED分析结果表明,HPX团簇的振动方式均为二面角扭转,ALP团簇则为键角弯曲和二面角扭转两种振动方式。EDA-FF数据和原子着色图表明,两者弱相互作用类型都是以静电相互作用为主、色散力为辅,ALP体系内氢键成键数目为HPX体系的两倍,并且色散作用都集中体现在与氢键直接作用的供体与受体原子上。研究结果表明,DFT与PED、EDA-FF分析方法相结合为结构相似的生物分子和分子间非键相互作用的深入研究提供了有价值的参考。
关键词 次黄嘌呤; 别嘌醇; 太赫兹时域光谱; 弱相互作用; 基于分子力场的能量分解分析
1 引 言
次黄嘌呤(Hypoxanthine, HPX, C5H4N4O)是肌苷的底物,同时也是核酸代谢的重要产物和重要的药物及药物中间体。别嘌醇(Apllopurinol, ALP, C5H4N4O)是HPX的同分异构体,自1966年ALP用于临床治疗以来,ALP一直是治疗各类痛风的首选药品,随后又发现了其新的应用领域。例如,某些肿瘤如淋巴瘤会导致高尿酸血症,因此ALP可用于治疗肿瘤溶解综合征[1,2], 并且其对预防由抗癌药引起的口腔炎和肌肉炎有益[3,4]; 初步结果表明,ALP也可以辅助治疗阿尔茨海默氏病[5]、躁郁症[6]和精神分裂症[7]。由于HPX和ALP独特的物理化学性质和良好的应用前景,对二者的研究一直是相关领域研究的热点。Hernández等[8]采用半经验量子力学、从头算量子力学和密度泛函理论(Density functional theory, DFT)研究了中性HPX和ALP在气相中的互变异构平衡和水溶液中的互变异构性。通过对最稳定的互变异构形式的检验,可以讨论互变异构对这些分子与黄嘌呤氧化酶识别与结合的功能意义。Ramaekers等[9]采用矩阵分离傅里叶变换红外光谱(Fourier transform infrared spectrometer, FTIR)技术、DFT和从头算方法研究了HPX化合物, 对比了RHF和DFT/B3LYP两种理论方法对振动频率的预测情况,并用MP2//DFT等4种方法预测互变异构体的相对能量和其与水的氢键配合物的相互作用能。Paragid等[10]采用从头算方法确定HPX对天然DNA碱基对的氢键偏好选择,并將选择性顺序与实验数据进行比较,其中,用DFT(Becke)计算出的几何结构最接近实验值,并且不需要太多的计算资源。Fernández-Quejo等[11]采用DFT-B3LYP杂化泛函和6-311++G(d,p)等4种基组,对溶液中次黄嘌呤两种主要的互变异构体HX/N7H和HX/N9H进行了振动分析。振动光谱为两种异构体在水溶液中共存的实验现象提供了理论依据,HX-d2光谱在1537 cm1处的IR吸收是由HX/N7H引起的,而HX溶液在1580 cm1处的Raman峰大致是由HX/N9H的振动引起的。Subbiah等[12]通过FTIR和FT-Raman光谱研究了室温下多晶样品HPX的分子振动。利用标准B3LYP/6-311+G**方法和基集组合,在全结构优化和基于DFT的力场计算之后,利用正则坐标分析解析光谱。Latosińska等[13]用1H-14N NMR-NQR双共振技术对HPX、ALP等进行了固态实验研究,用原子量子理论和DFT解释了在室温条件下测量的12个14N共振频率, 并鉴定了每种化合物中两类氮位N和NH。
虽然近年来研究人员借助红外光谱等技术和理论计算对HPX和ALP的互变异构现象和光谱现象开展了相关的理论与实验研究,并取得了一些成果, 但是,利用太赫兹时域光谱(Terahertz time-domain spectroscopy, THz-TDS)技术结合DFT等理论研究方法,对HPX和ALP的分子间相互作用(尤其是弱相互作用)的研究却鲜有报道。本研究应用THz-TDS技术测量了HPX和ALP样品的THz吸收光谱,并重点考察了HPX和ALP在THz波段的光谱吸收特性和色散特性。应用可在电子结构水平上准确研究分子间弱相互作用的DFT和可获得每种基团特征振动模式产生的贡献的势能分布(Potential energy distribution, PED)分析预测和解释THz实验光谱的吸收特性; 应用新颖灵活的基于分子力场的能量分解分析(Energy decomposition analysis based on forcefield, EDA-FF)定性可视化分析两种物质的弱相互作用成分。本研究从不同角度对HPX和ALP进行研究,为指导新药的设计与合成,以及疾病的治疗和新功能材料的设计提供了参考。
2 实验部分
2.1 实验装置
Z-3THz-TDS系统(美国Zomega公司),由超快飞秒光纤激光器、THz辐射产生装置、THz辐射探测装置和时间延迟控制系统四部分组成,实验系统原理详见文献[14]。此系统信噪比>70 dB,频谱分辨率>5 GHz。在室温环境进行实验时,需将密闭的THz光路充入干燥空气,使湿度<2%,以降低水分对THz波吸收的影响。
2.2 实验方法
2.2.1 样品制备 HPX和ALP(白色粉末,纯度99%, Sigma-Aldrich公司),使用前未进行进一步纯化处理。将样品置于YB-1A真空恒温干燥箱(天津盛达三合光学仪器有限公司)中干燥2 h, 去除水分。以3:1的质量比称取样品与聚乙烯粉末于研钵中,研磨混匀。在实验样品中添加聚乙烯,可使压片凝结得更牢固,并且聚乙烯在THz频段不产生吸收,不会对实验结果产生影响[15]。FW-4压片机(天津天光光学仪器有限公司)的压力调节为12 MPa左右,将每种样品研磨的混合物压制成直径13 mm、厚度1.2 mm的均匀薄片各3个。
2.2.2 数据处理 应用THz-TDS系统直接测得参考信号和样品信号的THz时域波形,经快速傅里叶变换后,获得其对应的频域谱; 根据文献[16,17]提出的Fresnel公式,提取频域谱中的吸收系数等光学参数,样品的吸光度可由公式(1)获得:
式中, ω是角频率, Es(ω)是样本信号的THz振幅,Εr(ω)是参考信号的THz振幅。最后,将吸光度数据从Matlab中导出制图,得到样品的THz吸收光谱。
3 理论计算和分析方法
3.1 密度泛函理论
HPX和ALP的构型搜索结果,均来自于英国剑桥大学的晶体结构数据库(The Cambridge Crystallographic Data Centre, CCDC)[18]。为了构造与物质晶体环境相符合的模型,并且适当描述分子间的相互作用,本研究依据Wright等[19]提出的模型,选取了单个分子由邻近6个分子围绕的团簇结构,分子文件均取自于已知的HPX和ALP晶体结构[20,21]。需要说明的是,这里使用团簇结构,而不是直接选用晶胞分析,主要是考虑到当前是对孤立体系进行模拟计算,如果仅用单个晶胞进行模拟,当分析某个分子与周围分子的相互作用能时,很可能该分子未被其它分子包围,造成与实际晶体环境不符。选定构型后,再结合适当的理论计算方法对HPX和ALP团簇进行光谱模拟,从实验结果与理论模拟的吻合程度判断并分析THz光谱的吸收特性。本研究选用对静电相互作用和交换互斥作用可定性描述的DFT方法,在杂化泛函B3LYP/6-311G**级别下,添加Grimme等[22]提出的D3色散矫正,以合理描述色散作用,对HPX和ALP体系进行几何构型优化,以寻找势能面上的能量最低点。在结构优化的基础上,对稳定的分子结构进行频率计算,并乘上基频校正因子以消除系统误差,结果无虚频,最后将光谱数据导出,在Origin软件中绘制出理论光谱图。
3.2 基于分子力场的能量分解分析
能量分解分析(EDA)是研究弱相互作用的重要方法,EDA可将片段间总相互作用能分解为多种有实际物理意义的成分,以便进一步考察体系内弱相互作用的本质。EDA-FF[23]相比于Morokuma、SAPT等其它主流基于波函数的能量分解方法,可以更灵活、便捷地考察分子内相互作用,并能通过VMD原子着色将弱相互作用的类型可视化。该方法的分子力场基本原理是根据静电作用和范德华作用(包括有排斥作用的交换互斥项和有吸引作用的色散项)定义,表达式如下:
其中,A和B为原子标号, r为原子间距离, q为原子电荷, ε为范德华作用势阱深度, R0为原子间非键距离,ε和R0是在分子力场内根据原子所处化学环境的类型定义的。静电作用仅通过原子电荷体现,当r=R0时,原子间范德华作用等于势阱深度。本研究利用Multiwfn支持的Amber力场,原子间范德华作用由各原子的范德华参数通过一定的规则计算得出,对每个原子定义非键半径参数R*, 原子间的非键距离R0也可由相应原子的非键半径加和得到,具体计算公式如下:
4 结果与讨论
4.1 实验与理论模拟结果分析
傅里叶变换红外光谱技术和拉曼光谱技术可提供分子骨架的振动和转动信息,X射线衍射技术能探测物质微观结构信息,而THz-TDS技术可将以上三者的优势相结合,有效地对样品组成的细微变化做出分析和鉴别,并准确捕捉THz低频段分子基团内在的振动和转动信息。图1为HPX在THz频段的实验光谱和理论模拟光谱图。由图1可见,理论模拟光谱的吸收峰出现在0.96、1.45和1.77 THz处,分别对应于THz-TDS实验吸收峰1.00、1.50和1.79 THz。图2为ALP的THz实验光谱与理论模拟光谱图。由图2可见,理论模拟光谱的吸收峰出现在0.75、1.01、1.38和1.78 THz处,依次对应THz-TDS实验吸收峰0.71、1.06、1.42和1.77 THz,其中,
0.71 THz处的吸收峰与文献[24,25]报道的一致。对比HPX和ALP的THz-TDS谱线可知,虽然两者互为同分异构体,但均具有各自特定的特征吸收峰,并且特征吸收峰的数目和整体走向大致相同。 同时, 图1和图2的理论计算吸收峰与 THz-TDS吸收峰之
间都存在偏移, 这是由于理论计算条件为0 K,而实验测量条件为室温(298 K)。另外,在HPX和ALP的THz-TDS光谱曲线中,有一些峰在理论模拟光谱中并未出现,这可能是由于理论计算是采用团簇结构模拟复杂的晶体结构环境,并且这两种物质均为有机物,分子间的相互作用对THz-TDS光谱曲线的吸收现象也产生了影响。总之,理论模拟计算的吸收峰都能与实验吸收峰吻合,表明本研究所选用的理论模型、基组和方法,可在当前的条件下很好地预测HPX和ALP的吸收特性。
4.2 简正振动分析
物质基团频率和特征吸收峰的红外光谱是其分子结构的反映,谱图中的吸收峰与分子中各基团的振动形式对应。当提交计算任务时,在Gaussian关键词中添加freq=intmodes。其次,借助GaussView软件对理论模拟结果的振动模式进行验证,分別截取了各吸收峰的分子簇集体振动模式(图3和图4)。为了更加完整地分析这些振动模式的属性,选择了可便捷地进行定性分析的振动分析软件PED,并将其与freq=intmodes结果进行综合,得出各吸收峰振动模式的归属结果,如表1所示。
由表1和图3可知,HPX团簇在0.96 THz处的振动模式表现为C76H19N86C94所在分子的二面角扭转; 1.45 THz处表现为H96O36C26C34所在分子的二面角扭转; 1.77 THz处表现为H43N49C45N47所在分子的二面角扭转。HPX团簇的振动方式均为二面扭转,这与团簇优化后的结构近似为平面密切相关。由表1和图4可知,ALP团簇在0.75 THz处的振动模式为N40H67N61所在平面的键角弯曲,以及N48N50H56N71和C72N71H56N50所在平面的二面角扭转; 1.01 THz处的振动模式为H47N48N50C55分子和H79N85N87C90所在平面的二面角扭转; 1.38 THz处的振动模式为N4H9N15所在分子、N17H21N38所在分子和H21N38C43所在分子的平面键角弯曲; 1.77 THz处的振动模式为N4H9N15N17分子和C2N4H9N15分子所在平面的二面角扭转。
4.3 弱相互作用分析与可视化
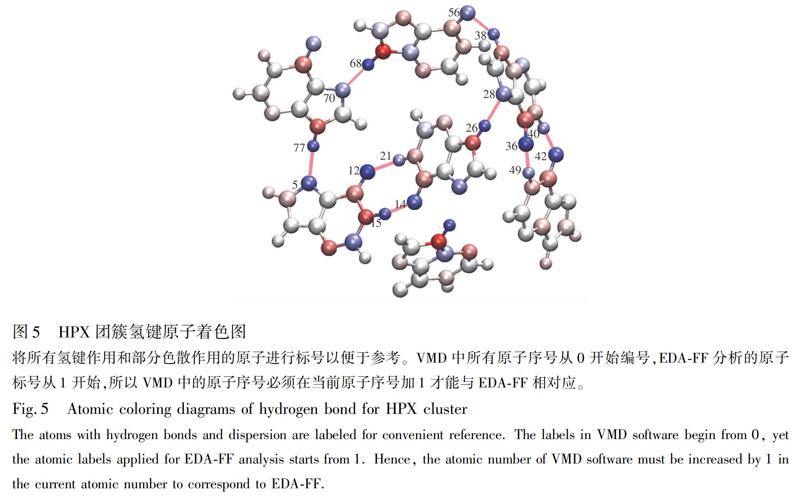
THz频段包含了丰富的光谱信息,尤其是有机分子,由于其转动和分子集体振动的跃迁,在这一频段表现出强烈的吸收和色散特性。尽管以上用PED分析方法对HPX和ALP的THz光谱吸收特性、分子官能团的振动和转动模式进行了分析,但要完整理解THz光谱信息,还须进一步鉴别体系内的弱相互作用成分。NCIpolt[26]程序在研究非共价相互作用(包含弱相互作用)方面取得了巨大成功,但其使用不便, 速度慢, 功能弱。因此,本研究选用便捷灵活、功能强大的波函数分析软件Multiwfn[27], 结合极具潜在应用价值的EDA-FF方法,对HPX和ALP的分子间弱相互作用进行分析。EDA-FF方法能够将体系内的弱相互作用分解为色散、静电和交换互斥作用,并以数据定性呈现其比例,再结合VMD软件,可将这些相互作用类型的本质以原子着色的方式展现各片段间的氢键和色散相互作用。
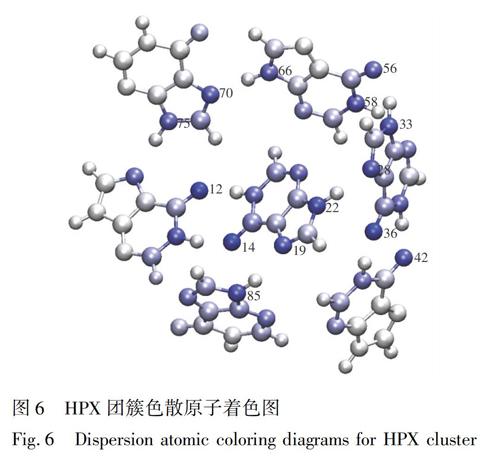
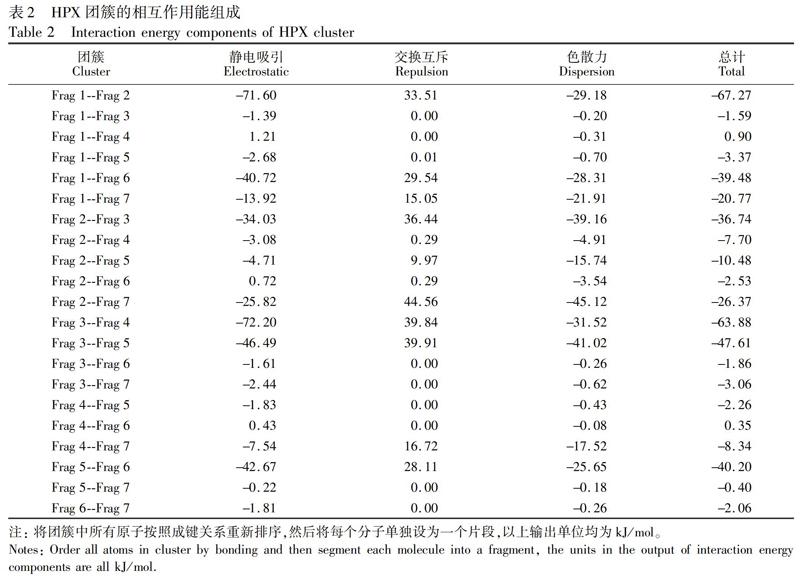
由EDA-FF分析结果(表2)可知,静电相互作用对HPX团簇的总结合能具有重要贡献,色散作用仅有少部分贡献,而交换互斥在数值上抵消了部分静电相互作用和色散作用。将结果数据导入VMD中,根据电荷属性及蓝-白-红(BWR)色彩变化方式着色,在氢键判据的范围内,绘制原子着色图(图5)。此着色方式表示原子电荷从最小值增加到最大值,因此颜色越蓝的原子具有越强的吸引力(其对总的结合能贡献值越负); 反之,越红的原子则有越显著的排斥作用,白色原子基本没有贡献。表2的数据表明,HPX是以静电作用为主导的弱相互作用体系,这归因于HPX体系内,氢键受体原子和与之作用的氢原子间形成了8对氢键,依次为:Frag 3的O37和Frag 4的H50、Frag 3的H41和Frag 4的O43、Frag 1的O13和Frag 2的H22、Frag 1的H14和Frag 2的O15、Frag 3的H39和Frag 5的O57、Frag 5的H69和Frag 6的N71、Frag 1的N6和Frag 6的H78、Frag 2的H27和Frag 3的N29。其中,两对静电高达70 kJ/mol的片段形成了两对氢键,分别为Frag 3和Frag 4,以及Frag 1和Frag 2。每条氢键的受体原子和与之作用的氢原子都是蓝色,表明其对结合的稳定性贡献极大。氢键供体原子颜色是红色,表明其不利于结合,这是因为氢键同时给供体原子和受体原子较大的相同符号的电荷,所以它们之间仍然存在明显的静电互斥作用。
表2的数据也表明,HPX体系总的色散作用在每个片段上的分布比较均衡。将色散数据导入VMD对原子进行着色,结果如图6所示。图6中, 颜色越蓝的原子,表示对片段间色散的吸引力越大,其中氢键供体原子和受体原子都是蓝色,如Frag 1的O13,Frag 2的O15、N20和N23,Frag 3的N34、N29和O37,Frag 4的O43,Frag 5的O57、N67和N59,Frag 6的N74和N71,Frag 7的N86等。色散贡献主要体现在氧原子和氮原子上,这是因为形成每对氢键的供体和受体原子之间的距离相近,并且所带电子数也相似。
有ALP的EDA-FF结果(表3)可知,此团簇的弱相互作用也是以静电主导,ALP团簇内氢键受体和与之作用的氢原子之间形成了16对氢键,如图7所示,依次为:Frag 1的H8和Frag 2的N15、Frag 1的O13和Frag 3的H35、Frag 1的O13和Frag 5的H61、Frag 1的H14和Frag 5的O67、Frag 2的N23和Frag 3的H39、Frag 2的H21和Frag 3的N29、Frag 2的H28和Frag 4的O48、Frag 3的N38和Frag 4的H43、Frag 3的H40和Frag 5的N57、Frag 3的O42和Frag 6的H75、Frag 3的H41和Frag 7的O95、Frag 3的O42和Frag 7的H89、Frag 4的H50和Frag 7的O95、Frag 5的N65和Frag 6的H82、Frag 5的H63和Frag 6的N71、Frag 6的H79和Frag 7的N85。其中Frag 1和Frag 5、Frag 2和Frag 3、Frag 5和Frag 6、Frag 3和Frag 7之间也形成了两对氢键。对比HPX的EDA-FF结论也知,HPX和ALP互为同分异构体,由于分子结构方面存在细微差别,导致几何优化后团簇的稳定构象和各体系内氢键的成键也有很大的差异。
表3的数据表明,ALP体系总的色散作用在每个片段上基本都有体现,如图8中颜色最蓝的原子:Frag 1的O13,Frag 2的N15、N17和N23,Frag 3的N29、N33、N38和O42,Frag 5的N57和N59,Frag 7的N85和O95等。由于氫原子所带的电子数很少,并且电负性小,在实际分子结构中容易失电子,所以产生的色散作用很小。将HPX和ALP体系内的色散作用对比可知,在相同的约束条件下,对色散作用起主要贡献的都体现在氢键的供体原子和受体原子上。
5 结 论
采用THz-TDS技术测量了HPX和ALP在室温环境下0.1~2.0 THz范围的THz吸收光谱,同时,应用DFT方法对THz实验光谱进行了理论模拟,并应用PED分析方法和EDA-FF方法分析了THz光谱的吸收特性和色散特性。理论模拟光谱能够较好地预测THz实验光谱中各吸收峰。分析结果表明,HPX团簇的振动方式均为二面角扭转, 而ALP体系则分为键角弯曲和二面角扭转两种振动方式。EDA-FF数据表明,静电相互作用为HPX和ALP体系的结合能提供了最主要的贡献,ALP较HPX体系各分子间的距离更近,因此形成了更多的氢键,但两者主导色散的原子类型相一致。研究结果表明,THz-TDS技术结合上述3种分析方法为研究结构相似的生物分子和分子间弱相互作用提供了一种有效的分析方法。
References
1 Maurer H S, Steinherz P G, Gaynon P S, Finklestein J Z, Sather H N, Reaman G H, Bleyer W A, Hammond G D. J. Clin. Oncol., 1988, 6(9): 1425-1432
2 Holdsworth M T, Nguyen P. Am. J. Health Syst. Ph., 2003, 60(21): 2213-2222
3 Saadeh C E. Pharmacotherapy, 2005, 25: 540-554
4 Nakamura K, Natsugoe S, Kumanohoso T, Shinkawa T, Kariyazono H, Yamada K, Baba M, Yoshinaka H, Fukumoto T, Aikou T. Anti-Cancer Drug, 1996, 7(3): 235-239
5 Lara D R, Cruz M R, Xavier F, Souza D O, Moriguchi E H. Int. Clin. Psychopharmacol., 2003, 18(1): 53-55
6 Fan A, Berg A, Bresee C, Glassman L H, Rapaport M H. Bipolar Disord., 2012, 14(2): 206-210
7 Buie L W, Oertel M D, Cala S O. Ann. Pharmacother., 2006, 40(12): 2200-2204
8 Hernández B, Luque F J, Orozco M. J. Org. Chem., 1996, 61(17): 5964-5971
9 Ramaekers R, Dkhissi A, Adamowicz L, Maes G. J. Phys. Chem. A, 2002, 106(18): 4502-4512
10 Paragi G, Pálinkó I, Van Alsenoy C, Gyémánt I K, Penke B, Timár Z. New J. Chem., 2002, 26(10): 1503-1506
11 Fernández-Quejo M, de la Fuente M, Navarro R. J. Mol. Struct., 2005, 744-747: 749-757
12 Subbiah J, Arivazhagan M. Indian J. Pure Appl. Phys., 2010, 48(2): 869-874
13 Latosińska J N, Latosińska M, Seliger J, agar V, Kazimierczuk Z. J. Phys. Chem. B, 2014, 118(37): 10837-10853
14 Chen T, Zhang Q, Li Z, Yin X, Hu F. Spectrochim. Acta A, 2018, 205: 312-319
15 ZHANG Liang-Liang, ZHANG Rui, HUANG Su-Xia, ZHANG Cun-Lin. Spectroscopy and Spectral Analysis, 2017, 37(2): 346-349
張亮亮, 张 锐, 黄素霞, 张存林. 光谱学与光谱分析, 2017, 37(2): 346-349
16 Dorney T D, Baraniuk R G, Mittleman D M. J. Opt. Soc. Am. A, 2001, 18(7): 1562-1571
17 Duvillaret L, Garet F, Coutaz J. IEEE J. Sel. Top. Quant., 1996, 2(3): 739-746
18 Groom C R. J. Chem. Inf. Model., 2011, 51(11): 2787
19 Wright A M, Joe L V, Howard A A, Tschumper G S, Hammer N I. Chem. Phys. Lett., 2011, 501: 319-323
20 Schmalle H W, Hanggi G, Dubler E. Acta Crystallogr. C, 1988, 44: 732-736
21 Prusiner P, Sundaralingam M. Acta Crystallogr. B, 1972, 28(7): 2148-2152
22 Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys., 2010, 132(15): 154104
23 Emamian S, Lu T, Kruse H, Emamian H. J. Comput. Chem., 2019, 40(32): 2868-2881
24 Song Q, Han P, Zhang X C, Zhang C, Zhao Y. J. Infrared Millim. Te., 2009, 30(5): 461-467
25 Kitagawa S, Mizuno M, Saito S, Ogino K, Suzuki S, Asada M. Jpn. J. Appl. Phys., 2017, 56(5): 58002
26 Johnson E R, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen A J, Yang W. J. Am. Chem. Soc., 2010, 132(18): 6498-6506
27 Lu T, Chen F. J. Comput. Chem., 2012, 33(5): 580-592
Study on Weak Interaction of Terahertz Spectra for
Crystalline Hypoxanthine and Allopurinol
ZHANG Qin, CHEN Tao*
(School of Electronic Engineering and Automation, Guilin University of Electronic Technology,
Guangxi Key Laboratory of Automatic Detecting Technology and Instruments, Guilin 541004, China)
Abstract The terahertz (THz) spectra of hypoxanthine and allopurinol samples were measured by THz time-domain spectroscopy (THz-TDS) in the range of 0.1-2 THz at room temperature. The geometry structures of these two samples were optimized using density functional theory (DFT). The low frequency vibrations of molecular group for these two samples were assigned using the potential energy distribution (PED) analysis. The spectral dispersion characteristics of samples were qualitatively analyzed by the energy decomposition analysis based on molecular forcefield (EDA-FF) method. PED analysis results showed that the vibration modes of the hypoxanthine cluster were all dihedral angle torsion, while allopurinol cluster were bond angle bending and dihedral angle torsion. EDA-FF data and atomic coloring diagrams indicated that the weak interaction types of the two clusters were dominated by electrostatic, while the amount of hydrogen bonds in allopurinol system were double compared with hypoxanthine, and the dispersion was mainly reflected on the donor and acceptor atoms that directly interacted with hydrogen bonds. The combination of DFT, PED and EDA-FF methods provided a valuable reference for the further study of intermolecular non-bonded interaction and biomolecules with structural similarities.
Keywords Hypoxanthine; Allopurinol; Terahertz time-domain spectroscopy; Weak interaction; Energy decomposition analysis based on forcefield
(Received 12 December 2019; accepted 19 May 2020)
This work was supported by the Natural Science Foundation of Guangxi Zhuang Autonomous Region, China (Nos. 2018GXNSFAA281167, 2018GXNSFAA138093) and the National Natural Science Foundation of China (No. 61841502).
2019-12-12收稿; 2020-05-19接受
本文系廣西自然科学基金项目(Nos. 2018GXNSFAA281167, 2018GXNSFAA138093)和国家自然科学基金项目(No. 61841502)资助
* E-mail: tchen@guet.edu.cn
