1号染色体三体、嵌合体和单亲二体的病例报道及文献综述
2020-08-13侯雅勤时盼来陈铎张弦叶梅汤冬娥郭辉冯宗辉谌燕孔祥东
侯雅勤 时盼来 陈铎 张弦 叶梅 汤冬娥 郭辉 冯宗辉 谌燕 孔祥东*
(1.郑州大学第一附属医院,河南 郑州 450052;2.怀化市妇幼保健院,湖南 怀化 418000;3.深圳市人民医院,广东 深圳 518020)
1号染色体是人类最大的染色体,片段大小为249.25Mb(hg38版本),包含大约8%的人类遗传信息和2.2亿多碱基。1号染色体上共包含3141个基因和991个假基因[1],基因的突变可导致多种疾病,如神经系统疾病、发育异常类疾病等。另外,1号染色体上的拷贝数变异(copy number variation,CNV)可导致1p36缺失综合征、桡骨缺失(thrombocytopenia-absent radius,TAR)综合征、染色体1q21.1重复综合征、染色体1q21.1缺失综合征等疾病。随着实验室检测技术的不断发展,低比例嵌合体、单亲二体(uniparental disomy,UPD)等也越来越多地被发现,给临床遗传咨询带来了更大的挑战。我们重点关注1号染色体完全型和嵌合型三体、UPD,参考以往文献、病例报道并结合笔者所在单位的数据,对1号染色体三体、嵌合体、UPD等信息进行综述。
1 1号染色体完全型三体和嵌合型三体
1.1 疾病概述 1号染色体三体综合征是一种非常罕见的染色体异常,核型为47,XX/XY,+1。该三体的发生机制一般为亲代生殖细胞减数分裂期1号染色体不分离导致。1号染色体三体对胚胎发育是致死性的,未见到在活婴中1号染色体三体的病例报道。
嵌合型三体通常发生在合子形成后的卵裂期或者胚胎正常发育的早期,可涉及包括性染色体在内的任何一条染色体。一部分三体细胞在分裂过程中启动三体自救(trisomy rescue)机制,将多余的1条染色体丢弃,恢复成正常的二倍体细胞,从而形成正常二体细胞和三体细胞的嵌合。目前为止,嵌合型1号染色体三体未见在新生儿中检出的报道。
1.2 临床特征 目前未见有1号染色体三体活产儿的报道。文献中在自然流产物中检出1号染色体三体的报道也不多。对于完全型1号染色体三体,迄今为止,只有1例植入前8细胞胚胎中[2]和2例临床确认妊娠后胚胎停止发育的绒毛中有检出[3,4]。本文中的3家医院(郑州大学第一附属医院、怀化市妇幼保健院、深圳市人民医院)产前样本(羊水、绒毛、流产物)未检出完全型1号染色体三体(表1)。因此,胚胎发育早期1号染色体三体是致死性的,胚胎在着床前后或发育早期无法继续发育,也有发育到妊娠4~5周,发生生化妊娠的报道[5,6]。
关于嵌合型1号染色体三体,综合3家医院共在2393例样本中仅只检出2例1号染色体嵌合型三体(表1),分别是在孕6周和9周流产胎儿中发现1号染色体三体嵌合,异常嵌合比例分别约为59%和46%(图1),均发生早期胚胎停育。由于孕周较小,均无超声或其他检测的结果。
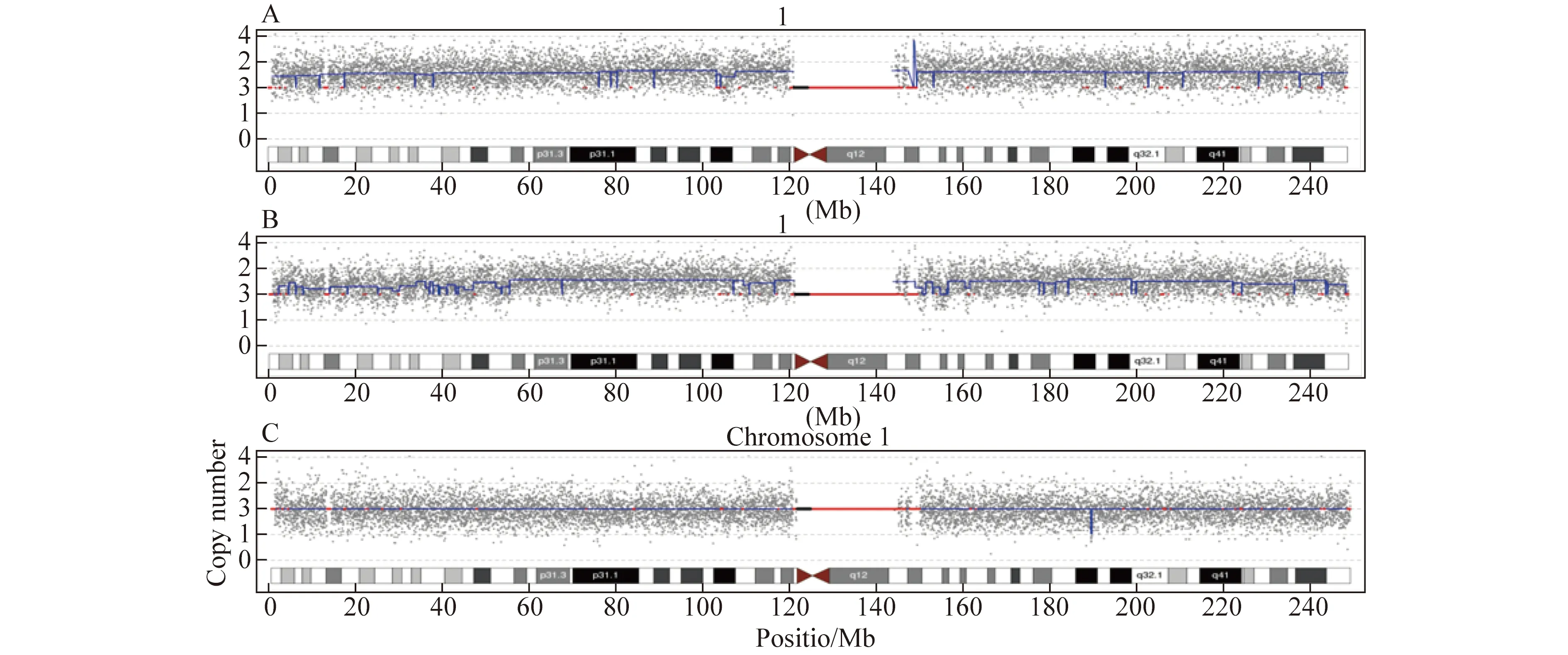
图1 2例流产物CNV-seq检测结果
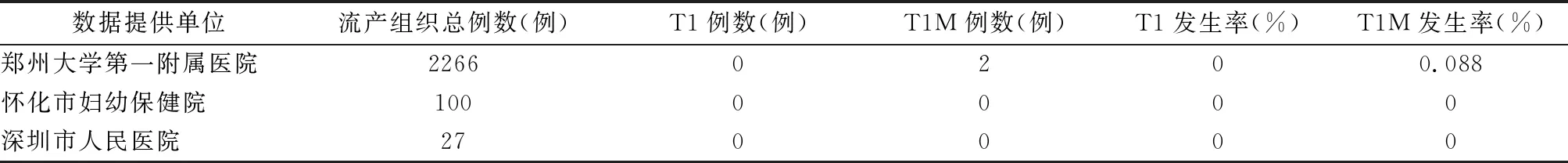
表1 流产组织中1号染色体完全型和嵌合型三体
1.3 再发风险评估及遗传咨询意见 1号染色体完全型和嵌合三体发生在受精卵形成后或者胚胎发育早期,再发风险很低,与正常人群相同。遗传咨询建议正常受孕,因不会发育到孕中期,可不建议在下次妊娠时进行针对性的产前诊断。
2 单亲二体
2.1 概况 UPD指个体的一对同源染色体全部或某个区域均来自于一个亲本,而不是分别来自父母双方。UPD发生频率约为1/2000~1/5000[7]。UPD主要是由于减数分裂不分离导致,按其形成机制可分为3类:①亲本某条染色体减数分裂Ⅰ或Ⅱ期不分离形成二倍体配子,然后与另一亲本正好缺少该染色体的配子形成合子;②三体细胞在分裂过程中丢弃了多余的一条染色体,成为正常二倍体细胞,留下的两条染色体均来自同一亲本(三体自救,trisomy rescue)[8,9];③正常的单体配子与缺少某条染色体的受精,由于配子的补足机制,该条染色体复制形成单体同源物合子(单体自救,monosomy rescue)[9]。
亲本基因的这种非孟德尔遗传可以导致罕见隐性疾病或由于印记效应导致发育紊乱[10]。由于UPD来源同一亲本的一条染色体,因此携带纯合的隐性遗传变异增加了患病的可能性。如果染色体携带印记基因且是胚胎生长发育所必须的,那么UPD也可以与宫内生长受限(fetal growth restriction,FGR)和其他发育异常相关[8,11],也可能会出现没有明显的表型UPD[12]。
2.2 1号染色体上的印记基因 目前Geneimprint网址(http:∥www.geneimprint.com/site/genes-by-species)收录了18个1号染色体上的印记基因,3个标记为“Imprinted”的为明确的印记基因,分别是DIRAS3、RNU5D-1、TP73,15个标记为“Predicted”的为潜在的印记基因,分别是NDUFA4P1、GFI1、FUCA1、BMP88、DVL1、WDR8、RPL22、PRDM16、PEX10、TMEM52、HSPA6、PTPN14、HIST3H2BB、OBSCN、OR11L1。DIRAS3和RNU5D-1为是1号染色体母系印记、父源表达,TP73为父系印记、母源表达。3个Imprinted明确的印记基因多与癌症相关,其中DIRAS3(OMIM 605193)在正常细胞中高表达,在乳腺癌中表达显著下调。用CpG去甲基化剂和(或)组蛋白去乙酰化酶抑制剂处理可以激活此抑癌基因的表达,在乳腺癌治疗中有指导意义[13]。
2.3 1号染色体UPD导致的疾病 目前未见1号染色体印记基因影响生长发育异常的报道,没有证据证实1号染色体上印记基因与胎儿发育相关[8,14,15]。已报道的1号染色体UPD病例都是因为隐性遗传致病基因纯合突变导致(表2),这些患者在产前检查均未表现出宫内生长受限等表型。Pulkkinen等[14]报道了第一例母源染色体1号染色体UP,Gelb等[15]报道了第一例父源1号染色体UPD,患者无宫内生长受限等症状,发病原因均为1号染色体片段性UPD导致隐性基因纯合突变。

表2 文献报道1号染色体UPD病例
2.3.1 遗传代谢相关疾病 Ponzi等[16]报道了1例1号染色体父源UPD病例。患者2~18岁身高生长曲线落后3个标准差,合并有低血糖症、肝肿大、肝脏疾病、肌肉疾病及心肌病。基因测序显示AGL基因c.3903-3904insA(p.N1304fsTer7)发生单核苷酸插入突变导致蛋白编码提前终止导致的糖原贮积病(glycogen storage disease,GSD)Ⅲ型。
Panzer等[17]报道了1例1号染色体UPD的男婴病例。患儿足月出生,出生身长、体重、头围均正常。体检发现患儿肌张力低下、面部特殊(前额突出、低鼻梁和鼻孔前倾)、会阴型尿道下裂、阴囊裂等异常。17α-羟基黄体酮在正常范围。核型检测结果显示患儿为46,XY。荧光原位杂交实验(fluorescence in situ hybridization,FISH)结果证实SRY阳性。染色体微阵列芯片分析(chromosomal microarray analysis,CMA)证实1号染色体为完全纯合状态的UPD。Sanger测序结果HSD3B2基因c.424G>A(p.Glue142Lys)纯合变异,患者诊断为类固醇3-β羟基类固醇脱氢酶类型Ⅱ(3β-HSD2)缺乏症,是先天性肾上腺增生症(congenital adrenal hyperplasia,CAH)的一种罕见类型。由于父母无法提供样本,因此无法确定突变的来源和UPD的亲代起源。
Aoyama等[18]报道了1例1号染色体父源UPD男性患者。患者3个月大时出现呕吐和神志不清,在4岁前有十几次低血糖危象,8岁时发育正常。多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)检测未发现存在外显子缺失。患者为HMGCL基因 c.242G>A(p.Trp81X)纯合变异导致的线粒体3-羟基-3-甲基戊二酰辅酶A裂解酶(hydroxymethylglutaryl-CoA lyase,HMGCL)缺乏。父母为非近亲结婚,父亲携带该位点杂合变异,母亲正常。
Beyec等[19]报道了1例1号染色体父源UPD病例。男性患者出生时身高体重正常,但体重迅速增长,婴儿期食欲不振但不伴畸形,精神发育正常,19岁出现性激素缺乏,诊断出睾丸发育不全,20岁卵泡刺激素(follicle-stimulating hormone,FSH)和促黄体生成素(luteinizing hormone,LH)水平正常。测序结果显示患者为LEPR基因 c.1870-1871insA(p.Asn624LysX21)导致的伴性腺功能减退性严重的食欲亢进早发性肥胖症。
Niida等[20]报道了1例1号染色体父源UPD的女性患者。该患者9个月前发育正常,此后发育迟缓(11个月会爬,13个月能站立等),共济失调。23个月龄出现不能独坐、睡眠障碍和四肢肌阵挛性抽搐。3岁时发现等电性脑电图和严重的脑萎缩。患者为PPT1基因发生移码突变c.20-47del28(p.Leu7Hisfs*21)而导致婴儿神经元类脂褐质病(infantile neuronal ceroid lipofuscinosis,INCL)。父母非近亲结婚,父亲为该位点的杂合突变携带者。
2.3.2 眼科疾病 Thompson等[21]报道1例51岁男性患者,通过17个多态性位点证实为患者1号染色体父源UPD。该患者为高级专业人员,核型正常,视网膜病发病于5岁(严重的视力丧失和眼球震颤),8岁时视力降到20/200,50岁时视野范围极小,无其他异常。因IVS6基因+5g>a纯合突变导致视网膜营养不良(retinal dystrophy),在儿童期已被确诊为Leber先天性黑曚(Leber congenital amaurosis)。
Fingert 等[22]报道1例女性患者,24个STR位点全部来自于父亲,为1号染色体父源UPD。15岁时视力为20/200 OD、20/150 OS,表现为典型视网膜特征。患者为ABCA4基因c.4139C>T(Pro1380Leu)纯合突变导致的Stargardt病。父亲携带该位点杂合变异。患者姐姐健康,遗传了父母两个人的等位基因。
2.3.3 免疫系统疾病 Dufourcq-Lagelouse等[23]报道了1例为母源UPD病例。患者为正常46,XY核型,足月出生,出生时体重3.20kg,无胎儿宫内生长受限、发育异常和其他畸形。患者是由于LYST基因c.2623delT(p.Tyr875Metfs*24)突变纯合变异导致Chediak-Higashi综合征(Chediak-Higashisyndrome,CHS)。除了CHS先证者,没有其他异常情况。患者父母非近亲结婚。患者母亲携带LYST突变,患者姐姐健康,继承了母亲和父亲的等位基因。
Roberts等[24]报道了1号染色体母源UPD病例。患者10岁骨髓白细胞CD45表达缺失,外周血淋巴细胞流式结果显示T细胞明显减少,血清IgM、IgA和IgE水平低到无法检测。二代测序结果显示患者为CD45基因 c.1618A>T(p.Lys540X)纯合突变导致的联合免疫缺陷综合征(severe combined immune deficiency mice,SCID)。研究还发现患者中性粒细胞、淋巴细胞和上皮细胞均为1号染色体UPD。
2.3.4 皮肤类疾病 首例报道的1号染色体母源UPD为长臂片段性母源UPD。患儿足月出生,除了Herlitz交界性大疱性表皮松解症外没有表现出明显的畸形。患儿为LAMB3基因c.727C>T(p.Gln243X)无义突变纯合,母亲为该位点杂合变异[14]。Fassihi等[25]也报道1号染色体父源UPD患者。该患者为女性,核型正常,出生早期就有广泛的皮肤起泡及甲周炎,5个月有鼻周有大面积创伤伴起疱,生长受限,无家族史。患者为LAMB3基因c.1903C>T(p.Arg635X)纯合变异导致的Herlitz交界性大疱性表皮松解症。Yasuko等[26]报道的1号染色体父源UPD患者出生时有皮肤和口腔黏膜起疱,8岁死于并发症。患者为LAMC2基因c.1776C>A(p.Cys553X)无义突变纯合导致的Herlitz交界性大疱性表皮松解症。父亲是该突变的杂合子,母亲正常。
Miura等[27]报道了1例1号染色体父源UPD,患者核型正常,对有害刺激无反应,无汗症,自残,智力迟缓,1岁时发育迟缓,但到2岁时身高体重发育正常。Sanger测序结果显示患者TRKA基因 c.1726delC(p.Arg548 fs)纯合突变导致的先天无痛无汗症。
2.4 再发风险评估及遗传咨询意见 UPD发生根源在于减数分裂不分离事件。1号染色体UPD患者主要是携带亲本常隐遗传病突变位点的纯合变异,再发风险低。从病例报道可见,先证者的兄弟姐妹多为正常。
