支化聚酯醚的一锅法制备及表征
2020-08-07左永康宋肄业杨宏军黄文艳薛小强蒋其民蒋必彪
左永康, 宋肄业, 杨宏军, 黄文艳, 薛小强, 蒋其民, 江 力, 蒋必彪,
(1. 江苏省绿色环保重点实验室 常州大学 材料科学与工程学院, 江苏 常州 213164;2. 常州大学 怀德学院, 江苏 靖江 214500)
1 前 言
聚己内酯(PCL)具有良好的生物相容性、降解性能、药物通透性能和力学性能,被广泛应用于降解包装、医疗器械和药物控释系统等领域,是最重要的环境友好型材料之一[1-3]。但是,线型 PCL 结晶度较高、亲水性较差导致其常温下降解慢、降解效果不理想[4-8]。为克服这一缺陷,科学家提出两种解决方案。第1 种为共聚[9-12]。例如,AHMED 等[12]报道的向PCL 中分别加入PEG 以及PLA 链段,成功赋予PCL更加优良的生物相容性与降解性能;HOQUE 等[13]利用PEG-PCL-PLA 三嵌段共聚物成功构建了具有可变机械强度和完全互联多孔网络结构的具有可控亲水性和可降解性的3D 聚合物支架;SUN 等[14]制备出超薄微孔PCL/PLA 膜,并测试了其与神经细胞和初级施旺细胞的相容性。其中聚乙二醇(PEG)具有独特亲水性及良好生物相容性,将PEG 引入PCL 中可在改善PCL 的亲水性能的同时,有效提高聚合物的降解性能;聚丙交酯(PLA)具有良好的生物降解性能,能被人体新陈代谢排出,将丙交酯与ε-己内酯(CL)共聚,不仅能保存PCL 优良的药物渗透性能,又能显著提高聚合物材料的生物降解性能[12-14]。但是,多嵌段型共聚物制备过程复杂,较难通过一步法或一锅法完成。第2 种改善PCL 降解性能的方法是在聚合物中引入多种拓扑结构。例如,在PCL 中引入支化结构能有效地破坏其链规整性,降低其结晶度,从而提高降解性能[15-17]。HUSKIC 等[15]提出一种用PCL 和BH40 进行接枝共聚的方法,可用于制备多臂星型PCL;宋肄业等[17]通过 CL 与甲基丙烯酸缩水甘油酯(GMA)共聚合成可降解大分子交联剂并将其应用于高内相乳液聚合制备出可降解聚合物多孔材料;XU 等[16]同时使用RAFT 和SCVP 两种聚合方式成功制备出结构可控的超支化聚己内酯。这些合成方法需引入特殊结构的第二单体共聚,其制备过程繁琐。为此,本文以CL、二丙烯酸新戊二醇酯(NPGDA)和乙醇胺(MEA)为原料,一锅法制备了以己内酯为主要结构单元的支化聚酯醚。所得共聚物中同时存在酯键、氨基、醚键等多种反应性基团和支化结构,不仅赋予了聚合物多样的功能性,同时聚合物的支化结构能破坏聚合物的链规整性,降低聚合物的结晶度,使聚合物具有较快的降解性能。
2 实验(材料与方法)
2.1 实验材料
乙醇胺(MEA),97%,Aladdin;二丙烯酸新戊二醇酯(NPGDA),> 89%(GC),TCI,加入氢化钙搅拌过夜,后经过减压蒸馏后使用,并在氮气氛围零度以下保存;ε-己内酯(CL),99%,Aladdin,加入氢化钙搅拌过夜,后经过减压蒸馏后使用,并在氮气氛围零度以下保存;四氢呋喃(THF),分析纯,国药集团化学试剂有限公司;THF,> 99.9%,Sigma-Aldrich;正己烷,分析纯,国药集团化学试剂有限公司;磷腈碱 t-BuP2,~2.0 mol·L-1in THF,Sigma-Aldrich。
2.2 聚合物的合成
在5 mL 圆底烧瓶内加入MEA (0.029 mL, 0.486 mmol),NPGDA (0.150 mL, 0.729 mmol),CL (0.778 mL,7.287 mmol),无水级THF,“冷冻-抽真空-解冻”循环3 次后,用氩气置换体系内残余气体3 次后密封,将其置于25 ℃ 恒温油浴锅中,经橡胶塞用微量进样器加入t-BuP2(24.290 μL, 0.049 mmol) 进行聚合反应。反应结束后,加入少量稀HCl 终止,并用THF 稀释,正己烷中沉淀,烘干后得到透明黏稠或膏状聚合物。
2.3 分析和表征
采用Waters 1515 三检测凝胶渗透色谱仪(triple detection gel permeation chromatography, TD-SEC)在35 ℃下对聚合物的分子量及分子量分布(PDI)进行表征。标样为窄分子量聚苯乙烯,流动相为 THF,流量为 1.0 mL·min-1,色谱分离柱为 4 柱串联(型号分别为:Styagel HR1THF 7.8 mm ×300 mm,Styagel HR3THF 7.8 mm ×300 mm,Styagel HR4THF 7.8 mm ×300 mm 和 Styagel HR5THF 7.8 mm ×300 mm),搭配 Waters 2414 示差折光检测器(refractive index detector,RI)、Wyatt 公司黏度检测器(ViscoStar-II viscometer)和Wyatt 公司多角度光散射检测器(MALLS,DAWN HELEOS-II) 3 个检测器。
采用Bruker ARX-400 核磁共振波谱仪(nuclear magnetic resonance spectroscopy, NMR)对样品结构进行表征。其灵敏度 > 100,分辨率 < 0.2 Hz。本实验中核磁共振氢谱使用氘带氯仿(CDCl3)作为溶剂,内标为四甲基硅烷(TMS),工作频率为400 MHz,磁场强度为7.05 T,常温测试。
采用DSC6000 差示扫描量热仪(differential scanning calorimeter, DSC)测量聚合物的热性能。DSC 类型为表面覆盖氧化铝的轻质炉体,采用热流式设计,传感器为镍铬合金样品平台,冷却附件为制冷或循环装置,标配双路数字质量流量控制器,扫描速率为升温20 ℃·min-1,扫描范围为 -60~120 ℃。聚合物的结晶度由其熔融焓与100% 结晶聚己内酯的熔融焓(139.5 J·g-1)的比值所得。
3 实验结果与讨论
3.1 支化聚酯醚的合成及表征
膦腈碱t-BuP2为非质子强碱,在理想情况下可同时催化MEA 上的氨基和羟基引发己内酯的开环聚合反应,形成含有羟基末端的星形聚己内酯(A3)。此外,研究发现 t-BuP2也能催化羟基与丙烯酸酯双键的Michael 加成反应[18-19]。因此,当己内酯的开环聚合体系中同时存在双丙烯酸酯类单体(例如NPGDA)时,生成聚己内酯的端羟基便能与双丙烯酸酯(B2)的双键发生Michael 加成反应[18-20],最终通过A3单体和B2单体的反应,得到以己内酯为主要结构单元的支化聚酯醚,反应如图1 所示。为得到目标产物,初定反应配方摩尔比[MEA]/[CL]/[NPGDA]/[t-BuP2]为 10/100/10/1,反应温度为 25 ℃。 在氩气氛围中加MEA、CL、NPGDA 以及溶剂THF 于反应瓶中,待体系混合均匀, 经3 次“抽真空-充氩气”步骤后加入催化剂t-BuP2。观察反应现象发现在加入t-BuP2之前,反应体系黏度却已经开始缓慢上升。这是因为即使在没有催化剂的情况下,少量MEA 上的氨基能与NPGDA 的双键发生Michael 加成反应,生成一端含有羟基,两端含有丙烯酸酯双键的化合物(图2 中AB2单体)。但是,这一反应并不影响反应产物的制备。这是因为己内酯单体仍可由AB2单体的羟基引发,最终参与到聚合物中。实验也验证了上述推断,当加入t-BuP2后,反应体系变黄,己内酯发生开环聚合反应,体系黏度迅速上升,不到1 min 转子就难以转动。由1H-NMR 测得NPGDA 双键转化率以及CL 的转化率分别高达96.9% 和96.2%。继续反应至6 h 后,在体系中加入HCl 终止反应并对产物进行纯化和表征,获得数均分子量与分子量分布分别为5.4×103g·mol-1和3.59 的支化聚合物。
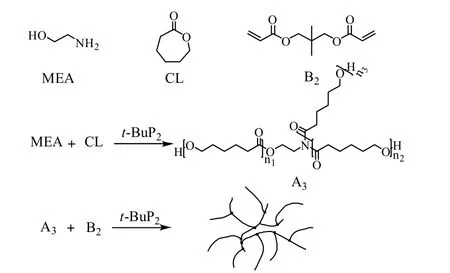
图1 由A3B2 结构生成支化聚酯醚机理图Fig.1 Mechanism of hyperbranched polyester ether synthesis from A3B2 structure
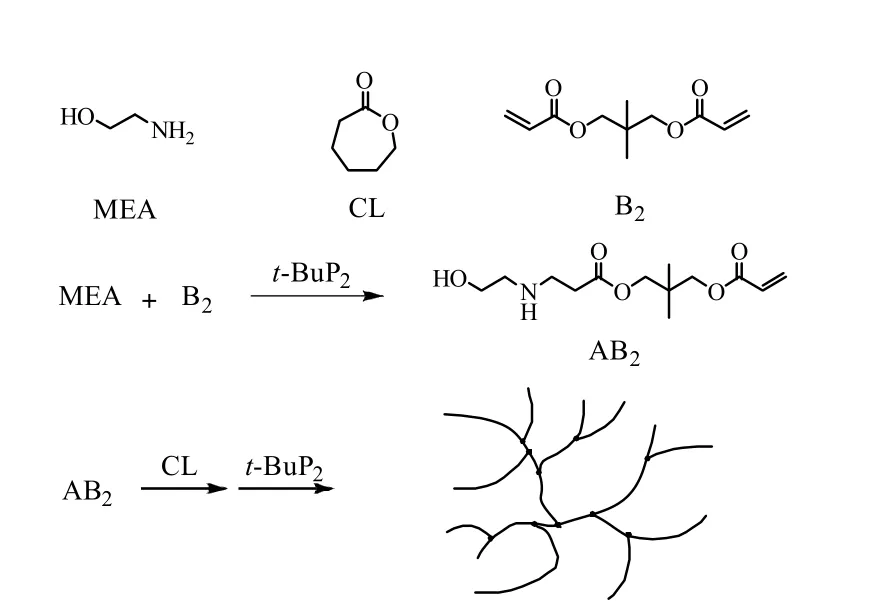
图2 由AB2 结构生成支化聚酯醚机理图Fig.2 Mechanism of hyperbranched polyester ether synthesis from AB2 structure
图3 给出了产物(h-PCL-1)的1H-NMR 图谱。图中,δ =4.00~4.13 (b)、δ = 2.26~2.40 (c)、δ = 1.52~1.74 (d)和δ =1.33~1.46 (e)分别对应于聚合物中己内酯结构单元上的5 个亚甲基的氢质子;δ = 0.86~1.02 (f)和δ = 3.82~3.95 (g)分别对应于NPGDA 结构单元上两个甲基和两个亚甲基的氢质子;δ =3.66~3.72 与δ = 2.51~2.62 (a)对应于NPGDA的双键进行Michael加成反应后所生成的两个亚甲基的氢质子;δ = 2.97~3.77 (h)的峰全都对应于MEA 结构单元上两个亚甲基的氢质子;MEA 上-NH2的两个氢都发生反应形成后,其上与 O 原子临近的亚甲基的峰会向低场偏移,出峰在δ = 4.39~4.45 (l)、δ = 4.46~4.56(m)和δ = 4.56~4.61 (n)处,这证明聚合物中存在支化结构。核磁结果说明MEA、CL 和NPGDA 都参与了聚合反应,生成了以己内酯为主要结构单元的支化聚酯醚。而且,对1H-NMR 图谱积分表明聚合物中MEA、CL 和NPGDA 3 种结构单元的摩尔比:1:9.93:1,与投料比接近。
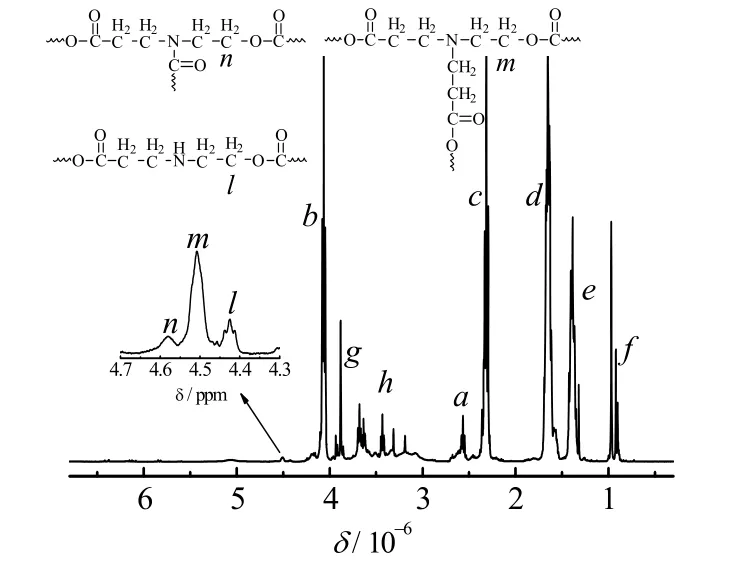
图3 h-PCL-1 的核磁氢谱图Fig.3 1H-NMR spectrum of h-PCL-1([MEA]/[CL]/[NPGDA]/[ t-BuP2] = 10/100/10/1)
为表征聚合物的支化结构,以乙二醇(EG)为引发剂,辛酸亚锡为催化剂,在120 ℃下合成了线型聚合物l-PCL-1 做对比。由SEC 测得其数均分子量为7.0×103g·mol-1,分子量分布为1.49。图4 给出了示差折光检测器测得的 h-PCL-1 和 l-PCL-1 流出时间曲线。从图中可以看出 l-PCL-1 呈窄分布单峰,而h-PCL-1 则呈现较宽的分子量分布,这可能是由于h-PCL-1 中存在不同支化度的组分导致其分子量分布变宽。

图4 示差折光检测器测得的h-PCL-1 和l-PCL-1 流出时间曲线Fig.4 Profiles of efflux time of h-PCL-1 and l-PCL-1 measured by a refractive index detector
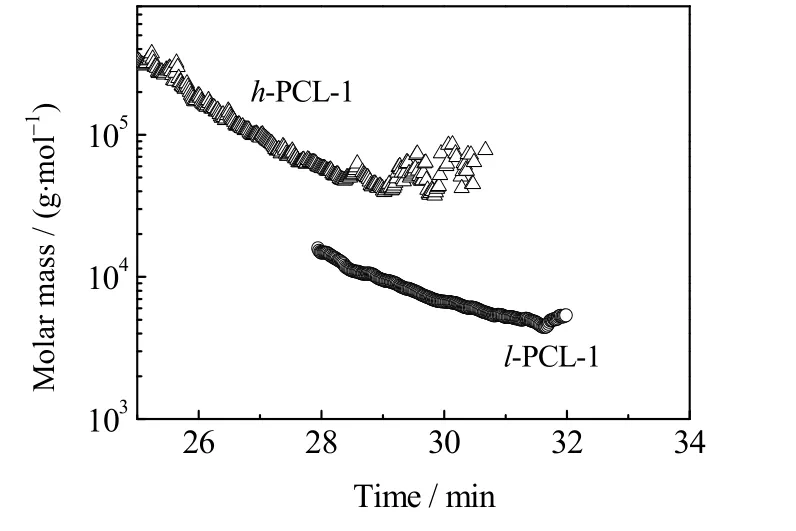
图5 h-PCL-1 和l-PCL-1 的分子量与流出时间曲线Fig.5 Profiles of molecular weight and outflow time of h-PCL-1 and l-PCL-1
图5 给出了由SEC 测得的聚合物h-PCL-1 和l-PCL-1 分子量与流出时间的关系曲线。从图中可以看出,在相同的流出时间,h-PCL-1 具有更高的分子量。例如,当流出时间在28 min 时,l-PCL-1 的分子量为 1.48×104g·mol-1,而 h-PCL-1 的分子量为 5.82×104g·mol-1,为 l-PCL-1 分子量的 3 倍,这证明 h-PCL-1具有紧凑的支化结构[21-22]。
图6 给出了聚合物h-PCL-1 和l-PCL-1 的流体力学半径与分子量的变化曲线。从图中可以看出,与l-PCL-1 相比,在相同分子量下h-PCL-1 具有更小的流体力学半径,这也证明h-PCL-1 具有支化结构。此外,从图中还可以看出随着分子量的增加,h-PCL-1 和 l-PCL-1 两者的流体力学半径差距越明显,说明h-PCL-1 的支化程度随分子量的增加而增加。
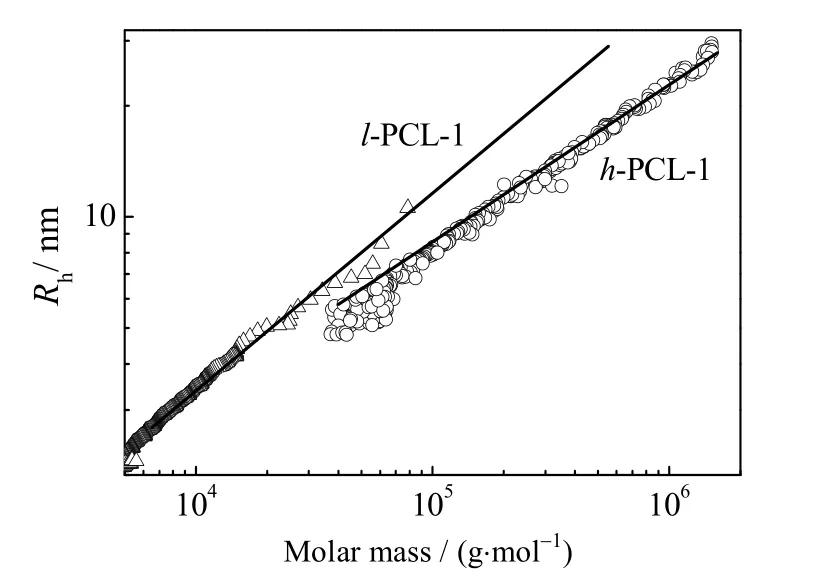
图6 h-PCL-1 和l-PCL-1 的流体力学半径和分子量曲线Fig.6 Profiles of hydrodynamic radius and molecular weight of h-PCL-1 and l-PCL-1
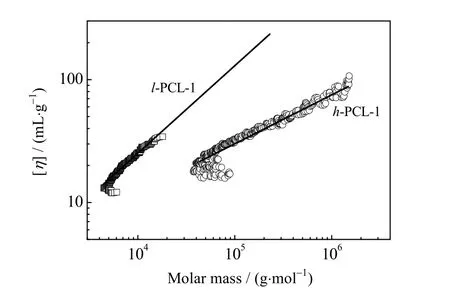
图7 h-PCL-1 和l-PCL-1 特性黏度随重均分子量变化的Mark-Houwink 曲线Fig.7 Mark-Houwink curves of h-PCL-1 and l-PCL-1
图7 分别给出了h-PCL-1 和l-PCL-1 的Mark-Houwink 曲线。从图中可以看出,在相同的重均分子量下,h-PCL-1 的特性黏度始终比l-PCL-1 低,这是因为支化聚合物独特的紧密堆叠的分子结构减少了分子间的相互缠结,从而降低了体系黏度,这也证明了h-PCL-1 确实具有支化结构。同样,从图中还可以看出随着重均分子量的变大,h-PCL-1 和l-PCL-1 的特性黏度差距越大。此外,Mark-Houwink([η]~Mα)曲线的斜率 α 可以用于推测聚合物的形状。α 值越小,聚合物支化度越大,对于支化聚合物,α 在0.3~0.5[22]。图中得出h-PCL-1 的α 值为0.46,远低于线型聚合物l-PCL-1 的α 值(0.69),这一结果再次证明合成了以MEA 为支化结构的聚酯醚[23]。
改变反应条件研究表明,当CL 的比例降低时(h-PCL-1 和 h-PCL-2,表1)所得聚合物Mark-Houwink参数 α 变小,说明降低CL 的比例,有助于提高聚合物的支化度,并且当其比例降低至10 倍于MEA时则极易交联成膜(例如h-PCL-3 和h-PCL-4),这也增加了其应用价值。当降低NPGDA 的含量时(h-PCL-5),NPGDA 双键转化率增高,并且支化度也会随之增加,而当降低MEA 的含量时(h-PCL-6),则双键转化率则会有所下降,并且支化程度变小,但因双键残留量多导致聚合物在反应后期易于交联。
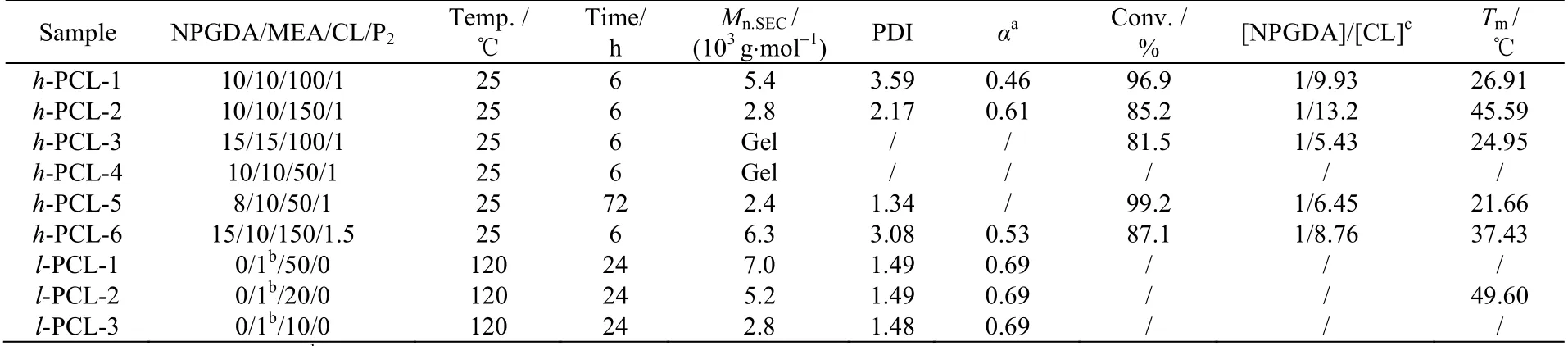
表1 杂化共聚制备支化聚己内酯Table 1 Preparation of branched polycaprolactone by hybrid copolymerization
3.2 支化聚酯醚的性能研究
以上研究结果证明MEA 可以引发NPGDA 和CL 进行杂化共聚反应生成支化聚酯醚。在此基础上,本实验还对聚合物的热性能进行了研究。图8 给出聚合物的差示扫描量热仪(DSC)曲线。结果表明,降低CL 比例 (h-PCL-1、h-PCL-2 和h-PCL-3,表1)导致产物支化度增加,结晶温度与结晶度降低。例如:线型聚己内酯(l-PCL-2)的结晶温度和结晶度分别为49.60 ℃ 和62.26%,而支化后的聚合物h-PCL-1 的结晶温度和结晶度则分别为26.91 ℃ 和24.63%。当产物过度反应交联后,则会显示出无结晶的现象。从图中还可以看出样品l-PCL-2 和h-PCL-2 出现两个熔融峰,这是因为聚合物在结晶过程中发生了不完善晶体的二次结晶。

图8 h-PCL-1、h-PCL-2、h-PCL-3 和 l-PCL-2的DSC 曲线Fig.8 DSC curves of h-PCL-1, h-PCL-2,h-PCL-4 and l-PCL-1
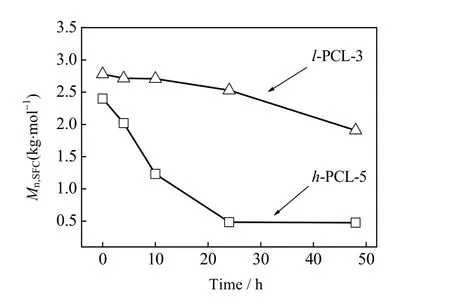
图9 h-PCL-5 和 l-PCL-2 的碱水降解曲线Fig.9 Profiles of alkaline degradation of h-PCL-5 and l-PCL-3
此外,本文还对数均分子量分别为2.4×103和2.8×103g·mol-1的支化聚酯醚(h-PCL-5)和线型聚己内酯(l-PCL-3)的降解性能就行了研究。图9 给出了聚合物在1% NaOH 水溶液中降解不同时间后聚合物的分子量变化曲线。从图中可以看出,反应48 h,h-PCL-5 的分子量减小了2.0×103g·mol-1,而l-PCL-3 的分子量仅减小了0.8×103g·mol-1。这是因为h-PCL-5 不仅具有较低的结晶度还具有醚键增加了其亲水性能,这赋予了它更加优异的降解性能[11]。
4 结 论
(1) MEA 上的-NH2和-OH 的3 个活泼氢都可以被催化产生反应,形成星状聚酯醚。 其后再由NPGDA的双键与活泼氢进行迈克尔加成反应将星状聚酯醚相互连接组合生成支化聚酯醚。当 CL 与支化单体MEA 投料摩尔比为1:15 时获得分子量为6.3×103g·mol-1,分子量分布为3.08 的支化聚合物。改变投料比,NPGDA 与CL 单体的转化率皆可接近100%,聚合物中结构单元的比例与投料比一致。
(2) 对聚合物的热性能以及水降解性能进行研究和表征,表明聚合物的结晶温度与结晶度相比于线型聚己内酯有着明显的下降,当CL 与支化单体MEA 投料摩尔比为1:10 时所获得的聚合物结晶度仅为24.63%,而再进一步增加支化单体 MEA 投料比时则聚合物会显示无结晶现象直至交联,且所合成的超支化聚酯醚在水中降解性能要明显优于线型聚己内酯。
符号说明:
Conv.=— NPGDA 双键转化率,%
Mn.SEC— 数均分子量,g·mol-1
PDI — 分子量分布
Tm— 结晶温度,℃
Xc— 结晶度,%
α — Mark-Houwink 参数
