液相色谱串联质谱法测定牛奶中黄曲霉毒素M1不确定度分析
2020-08-07王玉梅孙小杰南京市食品药品监督检验院
□ 王玉梅 刘 真 孙小杰 南京市食品药品监督检验院
黄曲霉毒素M1(A FM1)是含有二呋喃环的氧杂萘邻酮,是由黄曲霉毒素B1羟基化衍生而来[1]。AFM1对酸、光及热都很稳定,主要存在于动物的可食部分,比如蛋类、牛奶、肝脏等,其中以牛奶最为常见,目前已经成为乳制品质量安全风险评估的主要危害因素[2]。AFM1是目前发现的致癌性较强的毒素之一,它能够很大程度上破坏人及动物的肝脏组织,对人体产生肝肾等毒性,也存在致畸、致癌、致突变等风险,严重时会导致死亡[3]。本文参照JJF 1135—2005《化学分析测量不确定度评定》[4]、JJF 1059.1—2012《测量不确定度评定与表示》[5],JJG 646—2006《移液器检定规程》[6]和JJG 196—2006《常用玻璃量器检定规程》[7],依据GB 5009.24—2016《食 品 安全国 家标准 食品中黄曲霉毒素M 族的测定》(第一法 同位素稀释液相色谱—串联质谱法)[8]分析了牛奶中黄曲霉毒素M1含量检测的不确定度,以期为实验室质量控制提供有力的依据,同时也为其他真菌毒素类物质测定的不确定度分析提供参考。
1 材料与方法
1.1 仪器与试剂
Agilent 6470 串联四极杆液相色谱质谱仪(配制ESI 源);ME503T 电子天平(d=0.001 g);黄曲霉毒素M1标准品(10.0 μg/mL,纯度≥99%,阿 尔 塔);13C17—黄 曲 霉M1标 准品(BZYX483)(0.507 μg/mL,纯 度≥99.9%,ROMER)。
1.2 方法
1.2.1 标准溶液配制
准确移取黄曲霉毒素M11.00 mL于10 mL 容量瓶中,用乙腈溶解并定容,配制成1 μg/mL 的标准储备溶液。准确移取标准储备溶液1.00 mL 至10 mL容量瓶中,用乙腈稀释至刻度,得到100.0 ng/mL 的标准工作液。准确移取13C17—黄曲霉毒素M11.00 mL,用乙腈定容至10 mL,配制成50.7 ng/mL的同位素内标工作液。
将标准储备液逐级稀释,得到质量浓度为0.5、2.00、5.00、10.00、20.00 ng/mL 与25.00 ng/mL 的 黄 曲霉毒素M1标准工作液,其中内标的质量浓度为0.507 ng/mL,供高效液相色谱—质谱联用仪测定。
1.2.2 样品前处理
按 照GB 5009.24—2016[8]标 准 方法,称取4 g 混合均匀的试样(精确至0.001 g)于50 mL 离心管中,加入10 μL13C17—AFM1内标溶液(50.7 ng/mL)振荡混匀后静置30 min,加入10 mL 甲醇,涡旋3 min。置于4℃、5000 r/min下离心10 min,经玻璃纤维滤纸,将适量滤液转移至烧杯中,加40 mL水;将恢复至室温的免疫亲和柱内的液体弃掉,将上述样液移至50 mL注射器筒中,控制下滴流速为1 ~3 mL/min。待样液滴完后,往注射器筒内加入10 mL 水,并抽干亲和柱,加入2×2 mL 乙腈(或甲醇)洗脱亲和柱,收集全部洗脱液。在50 ℃下氮吹至近干,用初始流动相定容至1.0 mL溶解残留物,0.22 μm 滤膜过滤,待测。
1.2.3 色谱条件
色谱柱:Agilent Eclipse Plus C18 RRHD,粒度1.8μm,50×2.1mm(内径);流速:0.3mL/min;柱温:35 ℃;进样量:1μL;流动相:A—乙腈;B—0.1%甲酸/水,梯度洗脱程序见表1。
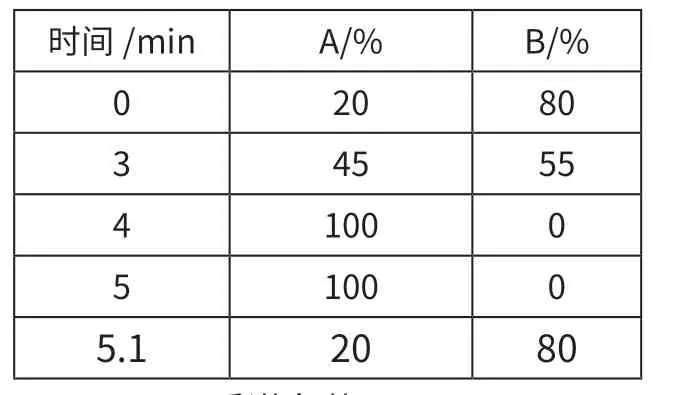
表1 梯度洗脱程序表
1.2.4 质谱条件
电喷雾离子源(ESI);正离子方式检测,检测方式为多离子反应监测(MRM);定量分析的离子对为:黄曲霉毒素M1母离子m/z 329.0,定量子离子m/z 272.7,定性子离子m/z 258.7;13C17—黄曲霉毒素M1母离子m/z 346.0,定量子离子m/z 287.9,定性子离子m/z 242.0。
1.2.5 数学模型的建立
被测物残留量X计算方法见式(1)。
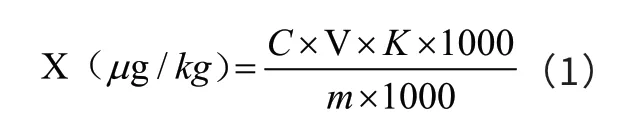
式(1)中:X ─试样中黄曲霉毒素M1的含量,μg/kg;C ─仪器检出目标物含量,ng/mL;V ─溶解试样或浓缩后所定容的体积,V=1.00 mL;m ─试样质量,g;K ─净化过程取样体积换算系数,K=1。
2 结果与分析
2.1 不确定度分量的来源分析
根据上述方法对样品进行分析时,测量的不确定度的主要有以下几个方面:①仪器检出目标物含量引入的不确定度。包含标准品纯度引入的不确定度,标准品称量、标准溶液稀释过程引入的不确定度,标准曲线拟合产生的不确定度。②样品前处理过程引入的不确定度。包括样品的称量、添加内标溶液、量取溶液体积以及加标回收率引入的不确定度。③样品重复测定引入的不确定度。
2.2 不确定度的评定
2.2.1 仪器检出目标物含量引入的不确定度Urel(1)
仪器检出目标物含量的不确定度主要由标准品纯度、标准系列溶液配置带来的不确定度。标准系列溶液配制又分为储备液的配制、储备液的稀释以及标准曲线溶液的配制。
(1)标准品纯度引入的相对标准不确定度Urel(1—1)
根据标准品证书提供的信息,黄曲霉毒素M1纯度不小于99%,按照矩形分布,由标准品纯度引入的相对标准不确定度为:
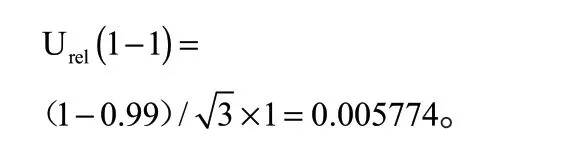
内标物纯度≥99.9%,内标对于标准品和样品的影响可相互抵消,因此由内标物纯度引起的不确定度不予考虑。
(2)标准系列溶液配置过程产生的不确定度Urel(1—2)
储备液配置及工作液稀释过程产生的不确定度Urel(p):准确移取黄曲霉毒素M11.00 mL 于10 mL 容量瓶中,用乙腈溶解并定容,配制成1 μg/mL的标准储备溶液。准确移取标准储备溶液1.00 mL 至10 mL 容量瓶中,用乙腈稀释至刻度,得到100.0 ng/mL的标准工作液。
在标准溶液配置及工作液稀释过程中,使用10 mL 容量瓶2 次,1 mL移液管2 次,按照JJG 196—2006《常用玻璃量器检定规程》的要求进行计算。
A 级 单 标 线1 mL(V1)移 液 管最大允许误差为:±0.007 mL,取矩形分布,其相对标准不确定度实验室温度为(20±5)℃,有机溶剂 体 积 膨 胀 系 数 为1.1×10—3/℃,1 mL 移液管由温度引起的体积不确定 度 为0.003175 mL,两者合成得到的相对标准不确定度为:

A 级单线10 mL(V2)容量瓶,最大允许误差为:±0.020 mL。取矩形分布,其相对标准不确定度为:0.01155 mL;10 mL 容量瓶由温度引起的体积不确定度为两者合成得到的相对标准不确定度为:
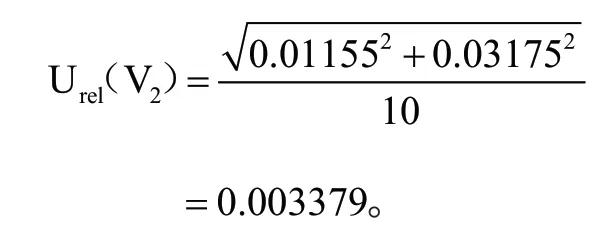
由此合成标准溶液配置及工作液稀释过程中引入的相对标准不确定度为:
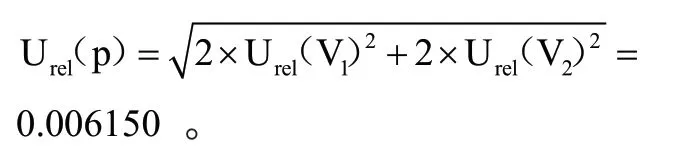
标准工作液稀释过程引入的相对标准不确定度Urel(x):标准工作液配置过程为分别精密移取5、20、50、100、200 μL 与250 μL 标准工作液至1 mL 容量瓶中,用初始流动相定容至刻度,得浓度为0.5、2.00、5.00、10.00、20.00 ng/mL 与25.00 ng/mL的标准系列溶液。移液器刻度误差参考JJG 646—2006《移液器检定规程》的要求,按照均匀分布处理,标准工作液配制过程引入的相对标准不确定度见表2。
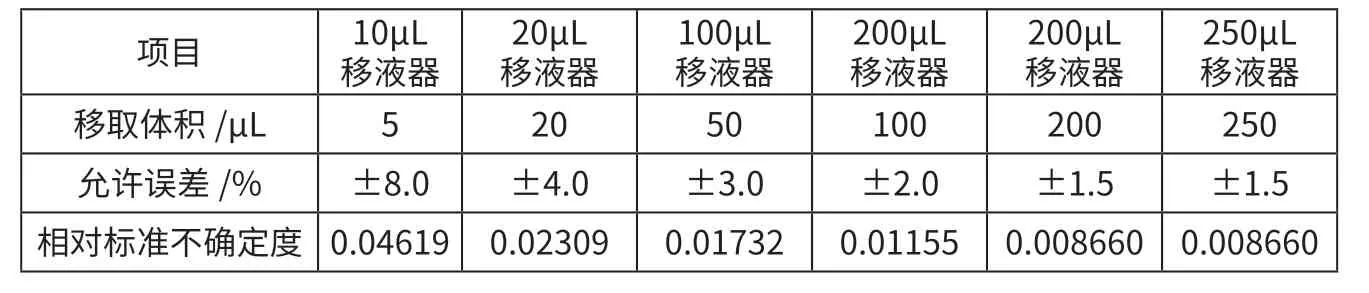
表2 标准工作液配制过程引入的相对标准不确定度
则由标准工作液配制过程引入的 相对标准不确定度为:

综上,标准系列溶液配置过程 中由各分量合成的相对标准不确定度为:
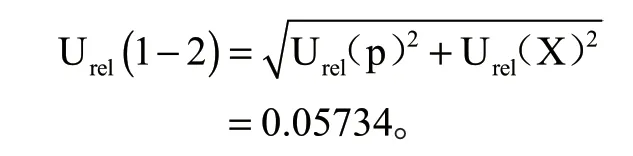
综上,由仪器检出目标物含量引入的相对不确定度为:
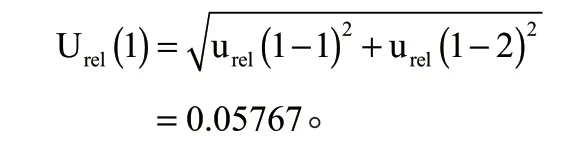
2.2.2 样品前处理过程引入的不确定度Urel(2)
(1)样品称量引入的不确定度Urel(2—1)
电子天平校准的最大允许误差为±0.001 g,称取食用油样品4 g,按矩形分布,引入的相对标准不确定度为:
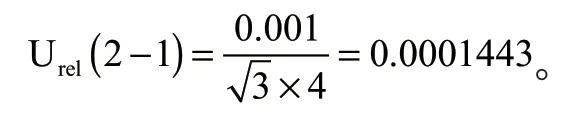
(2)添加内标溶液引入的不确定度Urel(2—2)
准确移取13C17—黄曲霉毒素M1储备液1.00 mL,用乙腈定容至10 mL,配制成5.07 ng/mL 的同位素内标工作液。采用200 μL 移液器准确量取5.07 ng/mL的13C17—黄曲霉毒素M1标准工作液100 μL,按 照JJG 646—2006《移 液 器检定规程》的要求,10 μL 移液器移取10 μL 时允许误差为±2.0%,取矩形分布,由移液器引入的不确定度为:
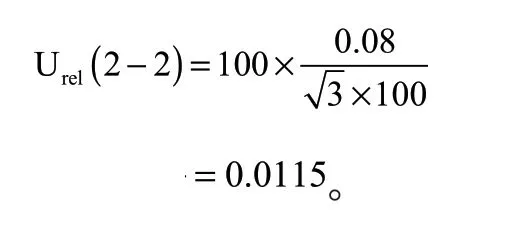
(3)量取溶液体积引入的不确定度Urel(2—3)
量取体积产生的不确定度主要是最终定容体积引入的不确定度。A 级1 mL单标线吸管最大允许误差为:±0.007 mL,取矩形分布,相对标准不确定度为:

(4)加标回收率引入的不确定度Urel(2—4)
对样品进行8 次加标回收实验,添加水平为6.25 μg/kg,回收率分别f 为101.2%、98.7%、96.5%、101.8%、103.0%与99.5%(见表3),平均回收率¯f 为99.3%,标准偏差为2.58%,由加标回收率引入的相对标准不确定度为:

综上,在样品前处理过程中,各分量合成的相对标准不确定度为:
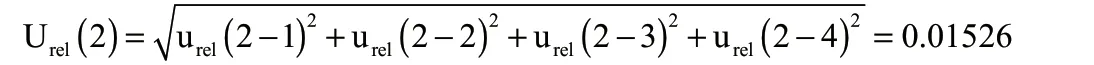
对被测样品溶液测量2 次,由回归方程计算得到样品中的黄曲霉毒素M 1 最佳估计值(Co),见表4。
2.2.3 样品重复测定引入的不确定度Urel(3)
对同一样品进行前处理和测定,重复8 次,计算得到样品中黄曲霉毒素M1含量分别为6.0092、6.0642、6.0313、5.9624、5.9770、6.0485、5.9953 μg/kg 与5.9776 μg/kg。平均值为6.0082 μg/kg,标准偏差为0.0377 μg/kg。由样品重复测定引入的相对标准不确定度为:

3 测量不确定度的评定与报告
3.1 合成相对标准不确定度
由上述计算得到的各不确定度分量的数据合成相对标准不确定度为:
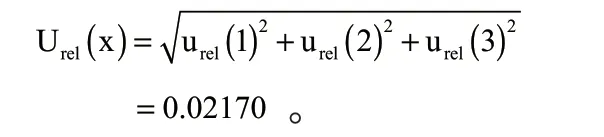
3.2 扩展不确定度
依 据JJF 1135—2005《化 学 分析测量不确定度评定》,在95.45%的置信水平下,取包含因子k=2,牛奶中黄曲霉M1的测量平均值为4.7342 ng/mL,结合称样量m 和定容体积V,计算得到样品中黄曲霉毒素M1的含量为1.1815 μg/kg,则扩展不确定度为:U=1.1815×0.02170×2=0.05128 μg/kg。
3.3 测定结果
按照该方法测定牛奶中黄曲霉毒 素M1的 含 量 为:X=(1.1815±0.05128)μg/kg;k=2。
4 结论
通过本次测定的不确定度评定的过程可以看出,使用GB 5009.24—2016《食品安全国家标准 食品中黄曲霉毒素M 族的测定》(第一法)定量分析牛奶中黄曲霉毒素M1含量时,其扩展不确定度为(1.1815±0.05128)g/kg,k=2,其中标准溶液及其工作液的制备对最终结果产生的影响最大,所以在实际检验工作中,需要提高检验人员在标准溶液配置方面的操作技能来提高结果的准确性。
