一种CRISPR-Cas9介导的拟南芥高效基因编辑系统的构建与应用
2020-08-04张成何明亮汪威徐芳森
张成,何明亮,汪威,徐芳森
(华中农业大学作物遗传改良国家重点实验室,武汉 430070)
0 引言
【研究意义】通过基因编辑手段获得基因敲除材料,对于研究基因在植物中的生物学功能具有重要的意义。而获得高编辑效率的CRISPR-Cas9系统能加速获得基因敲除材料的进程。【前人研究进展】在植物基因功能研究中,靶向基因编辑技术有着十分重要的作用,特别是在目标基因与其同源基因同源性较高的情况下很难通过其他途径获得想要的突变体。近些年,基于细菌免疫系统中CRISPR(clustered regularly interspaced short palindromic repeats)-Cas(CRISPR associated)的靶向基因编辑技术席卷了整个生物基因组研究领域[1-2]。常用的CRISPR-Cas9系统由两部分组成:带有核定位信号的Cas9和由21 bp靶序列(tracrRNA)的CRISPR(crRNA)构成的sgRNA(synthetic guide RNA)。其中,由sgRNA转录而成的非编码RNA与带有核定位信号的Cas9蛋白结合形成复合体,在PAM(protospacer-adjacent motif,Cas9系统中为NGG)位点上游3 bp位置切割DNA产生DNA双链切割(double-stranded breaks,DSBs),继而发生同源重组修复(homology-directed repair,HDR)或非同源重组修复(nonhomologous end joining,NHEJ),而容易出错的NHRJ修复方式导致在切割位点附近发生核苷酸的随机缺失,插入或者替换[1,3]。目前,CRISPR-Cas9系统已经成功在多种植物中得以应用,包括拟南芥、水稻、玉米、番茄、小麦、油菜和烟草等[4-11]。目前,在拟南芥中主要是利用农杆菌蘸花的方法,通过T-DNA插入到拟南芥卵细胞或者胚胎干细胞中的基因组获得拟南芥转基因植株。在之前的拟南芥CRISPR-Cas9研究中,主要是利用花椰菜花叶病毒(cauliflower mosaic virus,CaMV)35s启动子启动Cas9的表达,但发现拟南芥中的编辑效率较水稻下降很多,且发生基因编辑的T1代大部分为嵌合体。这主要由于35s启动子在植物的胚胎形成过程中表达量不高,而是在营养器官中高表达量的原因[8,11]。因此,为了提高拟南芥中基因编辑的效率以及遗传稳定性,选用合适的启动子启动Cas9的表达显得尤为重要。目前已经有一些研究人员使用了在拟南芥卵细胞中表达水平高的启动子来启动Cas9的表达,比如EC1.1/EC1.2启动子[12]、DD45和SPL启动子[13]、YAO启动子[14]、INCURVATA2启动子[15]、APETALA1启动子[16]以及RPS5A和WOX2启动子[17]。【本研究切入点】拟南芥CRISPR载体的编辑效率大多较低,且从T1代编辑株系的鉴定到T3代纯合突变且无Cas9材料的获得过程耗时费力,造成严重的时间、人力和资源的浪费。创建拟南芥高效的CRISPR编辑系统有利于加速基因敲除材料的获得,且节省时间和成本。【拟解决的关键问题】本研究通过构建一个高效CRISPR-Cas9系统用于拟南芥的基因编辑,并用拟南芥木葡聚糖内糖基转移/水解酶基因TOUCH 4(TCH4)对该基因编辑系统进行验证。采用DsRed2红色荧光筛选标记筛选阳性植株及无Cas9植株。通过对测序峰图的分析对编辑结果进行解码,以便降低获得稳定遗传的无Cas9突变体材料的时间、人力和财务成本。
1 材料与方法
1.1 材料
拟南芥为Columbia-0(Col-0)生态型,生长条件为长日照(16 h光照/8 h黑暗),环境温度为22℃。
1.2 拟南芥CRISPR-Cas9载体改造
拟南芥RPS5A编码一个核糖体蛋白,从胚胎发育早期到后期所有的生长发育阶段都表现为持续的高效表达[18-19]。使用的原始载体为pKSE401(由陈其军教授课题组提供),为了构建pRSE-WH载体,首先使用引物pKSE-F:5′-GCGGGACTCTGGGGTTCG-3′和pKSE-R:5′-GCGGGACTCTGGGGTTCG-3′,用宝生物公司的primer STAR MAX高保真酶扩增不包含35S启动子和潮霉素抗性的pKSE载体片段,同时以pBinGlyRed3的质粒为模板,用引物RED-F:5′-GCCAACATGGTGGAGGAGGAGTCCACCATGGT AGATCTG-3′和RED-R:5′-ACCCCAGAGTCCCGCT CAGGGTACCAGGAACAGGTG-3′扩增红光蛋白片段,利用Infusion(TaKaRa)将2个片段连接,转化后得到pRSE401载体。以pRSE401载体为模板,使用引物pRSE-F:5′-CTCGACCTCAACACAACATATA C-3′和pRSE-R:5′-GACCTGCAGGCATGCAAGC-3′,扩增得到不包含35S启动子的pRSE401载体片段。利用拟南芥基因组DNA为模板,使用引物AtRPS5A-F:5′-GCATGCCTGCAGGTCCTCAACTTTTGATTCGCT ATTTGC-3′和AtRPS5A-R:5′-TGTGTTGAGGTCGAG GGCTGTGGTGAGAGAAACAG-3′扩增得到AtRPS5A启动子片段,使用Infusion连接pRSE401载体片段和AtRPS5A启动子片段,得到pRSE-WH载体。
1.3 TCH4的CRISPR载体构建
拟南芥TCH4(AT3G11940,XTH22)属于木葡聚糖内糖基转移/水解酶(xyloglucan endotransglucosylase/hydrolase,XTH)家族中的一员[20],具有2种催化活性:1)内切木葡聚糖;2)将内切产生的木葡聚糖还原端连接到另一个木葡聚糖的非还原端。XTH一方面可以剪切细胞壁中的木葡聚糖交联网络引起细胞壁的松弛,一方面可以参与细胞壁新组分的组装,通过对细胞壁结构的调节参与对植物生长发育的调节[21-26]。利用CRISPR-P 2.0(http://crispr.hzau.edu.cn)对拟南芥TCH4设计2个靶点,靶点一序列为:5′-CCTTTCACTGCTTCTTACCG-3′,靶点二序列为:5′-GGATTGGAATCCAGAACCAG-3′。在引物的上游添加接头:5′-ATTG-3′,在引物的下游添加接头:5′-AAAC-3′。用于基因编辑第一个靶点的引物为:F:5′-ATTGCCTTTCACTGCTTCTTACCG-3′,R:5′-AAA CCGGTAAGAAGCAGTGAAAGG-3′。用于基因编辑第二个靶点的引物为:F:5′-ATTGGGATTGGAATCCA GAACCAG-3′;R:5′-AAACCTGGTTCTGGATTCCAA TCC-3′。采用25 µL退火体系(5 µL 10 mmol·L-1的上游引物,5 µL 10 mmol·L-1的下游引物,15 µL去离子水),退火程序为95℃ 3 min,95℃—16℃每20 s下降1℃,16℃保存,得到带有黏性末端的靶序列二聚体。酶切连接体系为靶序列二聚体2 µL、pRSE-WH 2µL、10×NEB T4 buffer 1.5 µL、BsaⅠ(NEB)1 µL、10×BSA 1.5 µL、T4连接酶(NEB,高浓度)1 µL,去离子水补充至15 µL。反应程序为37℃ 5 h,50℃ 5 min,80℃ 10 min。将连接产物转化大肠杆菌,送公司测序鉴定正确后,提取质粒转化农杆菌。
1.4 拟南芥转化以及阳性植株获得和筛选
采用沾花法转化拟南芥[27],收获T1代种子之后,筛选标记为红色荧光蛋白DsRed2,在绿色激发光下发射光为红色,使用体式荧光显微镜筛选阳性植株。
1.5 编辑鉴定
采用CTAB法[28]提取T1代阳性植株的叶片基因组DNA,使用引物:TCR-F:5′-ATGGCGATCACTTAC TTGCTTC-3′和TCR-R:5′-TACTCTCTCTATGCAGCT AAGCAC-3′进行PCR扩增,测序检测T1代植株编辑情况。
2 结果
2.1 拟南芥高效CRISPR-Cas9载体改造
首先,用DsRed2红色荧光蛋白替换了原始的潮霉素抗性基因(图1-A)。然后,利用拟南芥中RPS5A的启动子替换pKSE401载体中的CaMV35s启动子,从而达到在拟南芥胚胎发育早期进行基因编辑的目的。从拟南芥Col-0基因组DNA中扩增得到长度为1 659 bp的RPS5A的启动子,将其整合到pRSE-WH载体中(图1-A)。在pRSE-WH中,sgRNA由AtU6-26启动子启动,而在sgRNA和U6-26启动子中间包含2个BsaⅠ酶切位点,可直接一步将20 bp的靶序列连入载体。替换后的红色荧光蛋白可直接用于阳性植株的筛选以及后期无Cas9植株的筛选。
2.2 TCH4的CRISPR载体构建
TCH4的CDS全长为855 bp,基因结构包括2个外显子和1个内含子。使用CRISPR-P 2.0(http://crispr.hzau.edu.cn)对拟南芥TCH4设计2个靶点。第一个靶点位于第二个外显子上,第二个靶点位于第一个外显子上(图1-B)。在20 bp靶序列正向序列及反向互补序列的5′端添加接头序列,通过变性-复性形成带有黏性末端的二聚体,而pRSE-WH载体经过BsaⅠ酶切形成带有粘性末端的线性化载体片段,利用T4连接酶连接即可将靶序列整合到pRSE-WH载体中。将带有靶点的2个载体分别命名为TCR1和TCR2。

图1 CRISPR/Cas9载体图谱和TCH4的靶点示意图Fig.1 CRISPR/Cas9 vectors and target mapping of TCH4 gene
2.3 红光筛选
使用DsRed2作为筛选标记,用于阳性筛选。组成型表达的DsRed2蛋白,使得拟南芥种皮在绿光激发下发出红光,区别于其他阴性种子(图2)。将T1代红色的阳性种子播种(图2-A),收获得到T2代种子,大部分的T2代种子中,红光种子与非红光种子的比例近似3﹕1(红光种子与非红光种子分别为65和28颗,图2-B),符合分离定律。T2代挑选红色种子繁种得到T3代不同植株种子中,三分之一的几率得到纯合红色种子(图2-C)。而T2代非红光种子后代全部为非红光种子(图2-D)。
2.4 编辑检测
从T1代植株中挑选40株TCR1和19株TCR2,提取植株叶片DNA,对TCH4基因组片段进行PCR扩增,测序检测编辑结果。根据测序峰图手动解码编辑结果,结果发现,编辑类型可以分为4种:无编辑、纯合编辑、杂合编辑以及双等位编辑(图3)。在40株TCR1植株中,发生编辑的有32株,编辑效率达到80%,19株TCR2植株中,全部发生编辑,100%编辑效率。通过对不同的编辑类型进行统计,发现59株T1代阳性植株中,无编辑8株(13.56%)、纯合编辑9株(15.25%)、双等位编辑40株(67.80%)和杂合编辑2株(3.39%)(图4)。将每条染色体的编辑事件单独分析,118次编辑事件中,18次没有编辑,占比15.25%;45次单碱基插入,占比38.14%;1次多碱基插入,占比0.85%;11次单碱基缺失,占比9.32%;43次多碱基缺失,占比36.44%(表1)。其中多碱基缺失中,从几bp到几十bp均有发生,甚至几百bp缺失。在单碱基插入事件中,TCR1中只存在2种事件,4次A碱基插入和28次T碱基插入,而TCR2中4种碱基插入均有发生,5次A碱基插入,2次T碱基插入,1次C碱基插入和5次G碱基插入。

图2 阳性植株的筛选Fig.2 Screening of positive plants
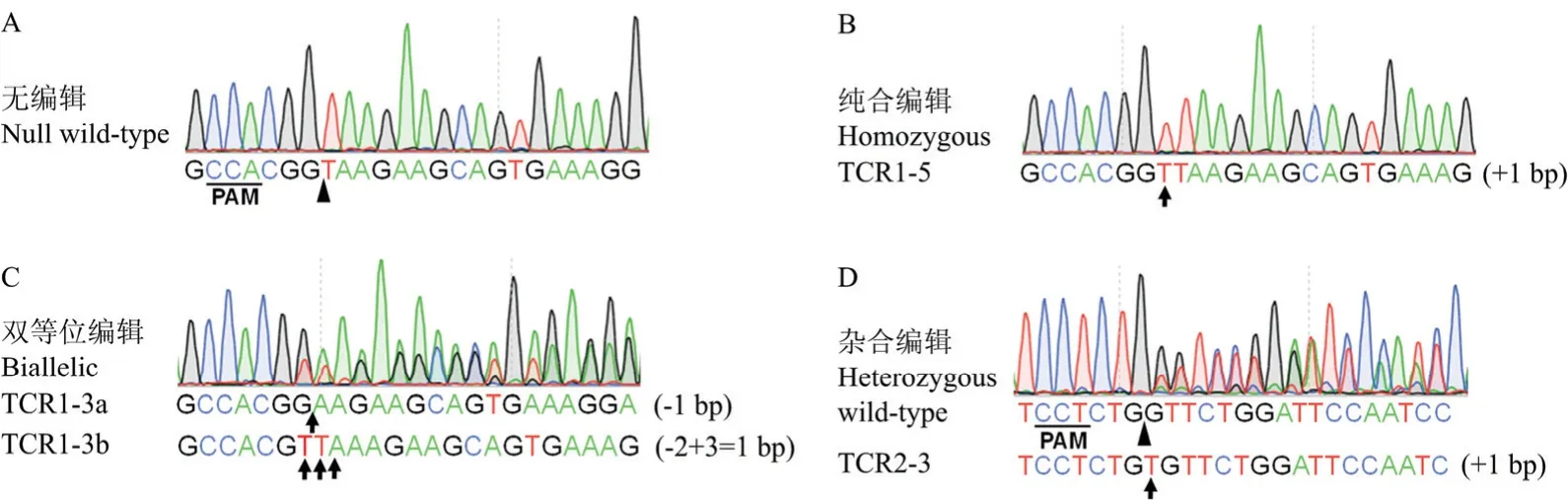
图3 T1代植株的编辑类型Fig.3 Genotypes of the edited T1 plants
2.5 T2代编辑分析
在T1代发生纯合编辑以及双等位编辑的株系中选择了无红光种子进行繁种,并对T2代植株编辑情况进行测序检测。结果显示,TCR1-27株系T1代检测结果为T碱基插入的纯合编辑,所测序的6个T2代单株均为纯合的T碱基插入。同样,TCR1-10和TCR1-11的T1代均为双等位编辑,所测序的6个T2代单株均显示2条染色体中的突变都成功地遗传到后代中(图5)。

图4 T1代植株各编辑类型所占比例Fig.4 Proportion of editing types in T1 plants
3 讨论
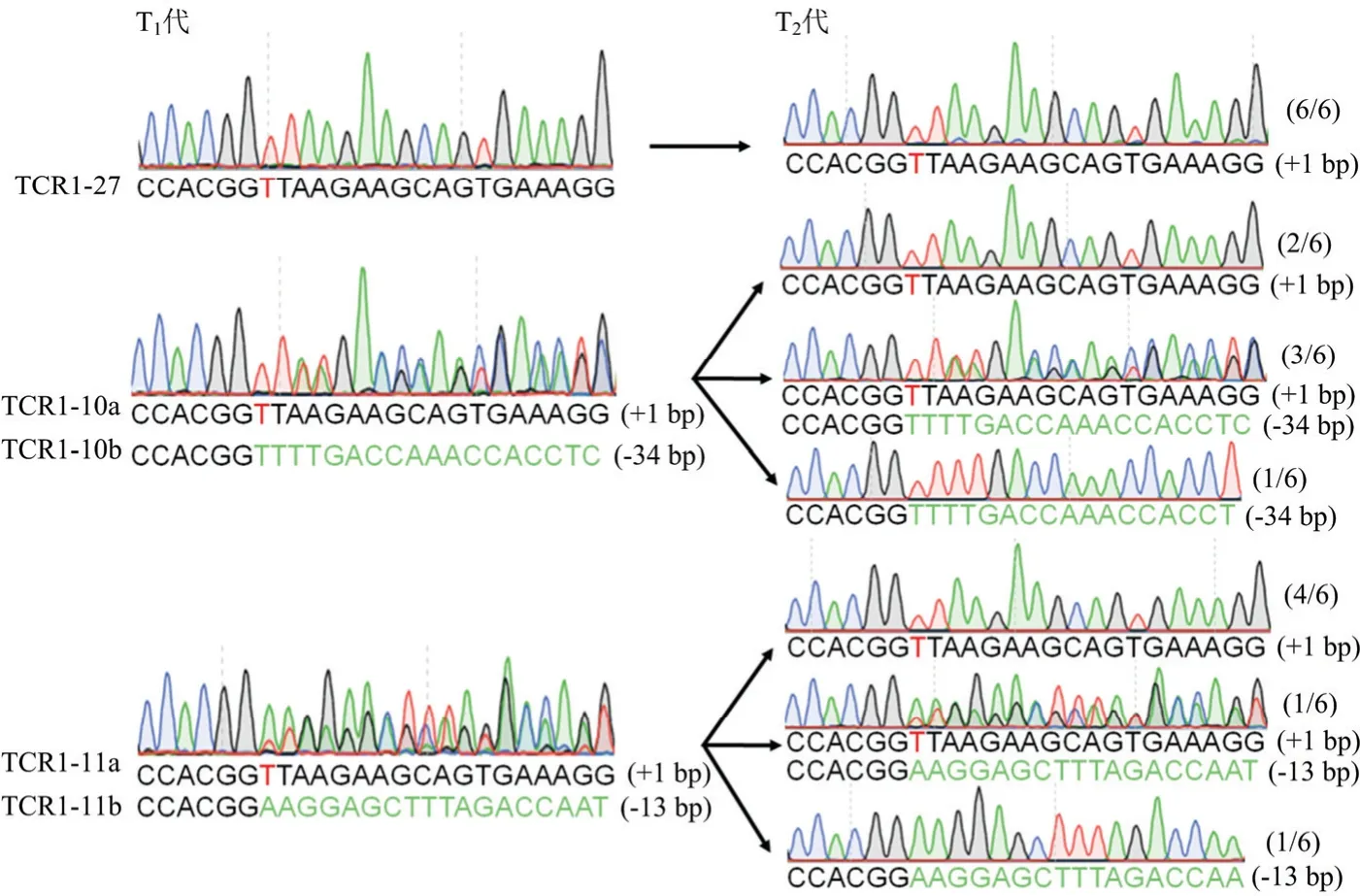
图5 T1和T2代株系间的突变遗传Fig.5 Mutation inheritance in T1 and T2 generation
以CRISPR为代表的基因编辑技术已广泛应用于农作物基因功能验证和农作物遗传改良。目前,已研发多种CRISPR载体运用于单子叶和双子叶植物的基因编辑[11-14,17,29]。植物营养遗传实验室尝试了使用载体pKSE401在拟南芥和油菜中运用CRISPR技术对拟研究的候选基因进行基因编辑。但是发现在CRISPR实际操作中存在的两个问题,一编辑效率低;二从T1代阳性植株筛选到T3代稳定遗传株系的获得,工作量大,成本高。而前者是主要的问题,因为一个高效的编辑系统能够很大的降低后期的工作量和成本。植物营养遗传实验室前期的工作中使用载体pKSE401进行甘蓝型油菜和拟南芥的编辑,其编辑效率较低,造成严重的时间、人力、资源的浪费。于是尝试了对实验室原有的CRISPR载体进行改造,试图提高编辑效率。与一些植物采用外植体侵染-再生转化植物不同的是,拟南芥遗传转化采用的是沾花法,农杆菌侵染的是雌性生殖器官中的胚囊。常用的35S启动子在胚囊中或者在整个胚胎发育过程中表达量并不高,而这很有可能导致了拟南芥中编辑效率的下降。因此,本研究采用了一个在胚胎发育过程中持续高表达的RPS5A启动子替换35S启动子。
对TCH4的编辑结果显示,RPS5A启动子启动Cas9的系统确实有着很高的效率,测序的59株转基因阳性植株中编辑效率达到86%。而不同的靶点序列的编辑效率也有一定的差异,其中靶点一的编辑效率为80%,而靶点二的则是100%编辑。目前,CRISPR在水稻中的运用较好,编辑效率可以超过81%[11],在拟南芥中的编辑效率大多不超过40%[7,11,16]。而本研究改造的CRISPR编辑效率可高达86%。高的编辑效率可以很容易得到各种不同的突变类型,发生最多的是单碱基插入,占38.14%;其次是多碱基缺失,占36.44%;再次是单碱基缺失,占9.32%;而多碱基插入发生概率较低,118次编辑事件中只发生了一次。在这些不同的突变类型中,发现不同的靶点序列似乎对不同的突变类型有一定的偏好性。在TCR1的单碱基插入中,只发现了A/T插入,而且T插入占88%。但是TCR2的单碱基插入则4种碱基插入均有发现。导致这样的结果可能与产生DNA双链切割位点的碱基类型有关。TCR1的DNA双链切割位点碱基为G-T,而TCR2的DNA双链切割位点碱基为G-G。

表1 TCH4 CRISPR T1代植株的编辑类型及比例Table 1 Mutation patterns and proportions TCH4 edited T1 plants
DsRed最早是从香菇珊瑚(Discosoma striata)中分离得到的一种红色荧光蛋白[30]。DsRed的发射光谱峰值为583 nm,激发光谱峰值为558 nm。DsRed在应用过程中被发现存在一些问题,包括成熟缓慢,易形成四聚体,有一定毒性。第二代DsRed2的氨基末端进行了一些突变改造,使得其组织蛋白凝集和毒性下降,荧光基团成熟时间变短。DsRed2不仅可以像GFP一样进行活体检测和连续观察,而且能消除植物本身的背景干扰,甚至在白光条件下也能检测到红光,因此,DsRed2可以作为一种可视标记应用于植物遗传转化[30-31]。在pRSE-WH中,采用DsRed2这种可视筛选标记。如果Cas9一直在植物中表达,它一方面可能会继续对靶位点进行切割、编辑;另一方面脱靶的可能性也一直存在。因此,最终希望得到无Cas9且稳定遗传的突变体。而Cas9基本上是与DsRed2连锁的,所以通过筛选无红光种子就能得到无Cas9的株系。在拟南芥CRISPR系统中,常用的抗生素筛选标记不适用于T2代无Cas9株系的筛选,而只能对T2代植株提取DNA,通过PCR扩增进行鉴定,理论上这会增加很大的工作量。而使用DsRed2可以很轻易的从T2代种子中筛选得到无Cas9的种子,并且能够保证T3代种子中无Cas9的稳定遗传。所以,在拟南芥CRISPR系统中DsRed2要明显优于常用的抗生素筛选。此外,常用的解码编辑结果的方法为对PCR产物进行克隆测序,并增加测序的单克隆数量获得编辑的结果,这无疑是最可靠的方式之一,但这会增加大量的时间、人力和物力的消耗。本研究在了解解码的机制后进行手动解码:对目标基因的基因组片段进行PCR扩增,测序检测编辑结果会出现2种峰型:单峰和套峰。其中,单峰表明扩增的PCR片段是单一的,可以很容易得到其中的DNA序列信息,而这种单峰的结果包括没有发生编辑和纯合编辑;套峰意味着PCR产物存在2个或者多个不同的DNA片段,会导致没有那么容易获得其中不同的DNA序列信息。可以通过以下几点去解码套峰中包含的信息:1)PCR过程中会导致不同DNA片段含量不一致,进而测序中表现出不同强度的信号值,可以通过这种信号强度的差异将不同的信息区分开;2)由于CRISPR-Cas9是基于DNA双链切割进而发生DNA修复产生的编辑,所以它的特点是编辑会发生在切割位点附近或者向外扩散,而未编辑的位点则应该与参考序列一致,因此,即使在套峰中2个信号强度相近的DNA片段,依旧可以通过碱基间的序列信息推断出其序列,解码出其中一条信息之后,套峰的信息减去其中已知的信息,就能得到另一条的序列信息。基于以上两点,对T1代阳性植株的测序信息进行解码,选择理想的编辑株系进行繁种,并在T2代中进行测序验证,其结果等同于在T1代中进行克隆测序,且节省了大量的时间、精力和成本。
4 结论
构建了一个适用于拟南芥中基因编辑的高效CRISPR载体pRSE-WH,能够简便地获得无Cas9且稳定遗传的T3代突变体。
致谢:中国农业大学陈其军教授提供了原始pKSE401载体;华中农业大学杨光圣教授课题组提供了体式荧光显微镜;华中农业大学汪社亮博士和英国The James Hutton Institute的Philip John White教授对文章的修改,在此表示感谢。
