GPS 5.0:An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins
2020-07-29ChenweiWangHaodongXuShaofengLinWankunDengJiaqiZhouYingZhangYingShiDiPengYuXue
Chenwei Wang ,Haodong Xu ,Shaofeng Lin ,Wankun Deng ,Jiaqi Zhou ,Ying Zhang ,Ying Shi ,Di Peng ,Yu Xue ,2,*
1Key Laboratory of Molecular Biophysics of Ministry of Education,Hubei Bioinformatics and Molecular Imaging Key Laboratory,College of Life Science and Technology,Huazhong University of Science and Technology,Wuhan 430074,China
2Huazhong University of Science and Technology Ezhou Industrial Technology Research Institute,Ezhou 436044,China
KEYWORDS Protein phosphorylation;Protein kinase;Group-based Prediction System;Kinase-specific phosphorylation site;Dual-specificity kinase
Abstract In eukaryotes,protein phosphorylation is specifically catalyzed by numerous protein kinases(PKs),faithfully orchestrates various biological processes,and reversibly determines cellular dynamics and plasticity.Here we report an updated algorithm of Group-based Prediction System(GPS) 5.0 to improve the performance for predicting kinase-specific phosphorylation sites(p-sites).Two novel methods,position weight determination(PWD)and scoring matrix optimization(SMO),were developed.Compared with other existing tools,GPS 5.0 exhibits a highly competitive accuracy.Besides serine/threonine or tyrosine kinases,GPS 5.0 also supports the prediction of dual-specificity kinase-specific p-sites.In the classical module of GPS 5.0,617 individual predictors were constructed for predicting p-sites of 479 human PKs.To extend the application of GPS 5.0,a species-specific module was implemented to predict kinase-specific p-sites for 44,795 PKs in 161 eukaryotes.The online service and local packages of GPS 5.0 are freely available for academic research at http://gps.biocuckoo.cn.
Introduction
Protein phosphorylation plays a critical role in almost all of biological processes and greatly expands the proteome diversity.By covalently attaching phosphate moieties to serine,threonine, and/or tyrosine residues in a dynamic manner,phosphorylation can reversibly change the structure,enzymatic activity,and subcellular trafficking of proteins[1,2].In eukaryotes,phosphorylation reaction is differentially and specifically catalyzed by numerous protein kinases(PKs),and each PK only modifies a limited subset of substrates to ensure the signaling fidelity[3-5].Aberrances in either PKs or phosphorylated substrates are highly associated with human diseases such as cancer[6,7].Therefore,the identification of kinase-specific phosphorylation sites(p-sites)is fundamental for understanding the regulatory mechanisms of phosphorylation.
Besides experiments,bioinformatics provides an alternative means for computational prediction of potential PK-specific p-sites from protein sequences[8-15](Table S1).In 2004,we developed a novel algorithm,group-based phosphorylation site predicting and scoring(GPS)1.0,based on the hypothesis that similar short peptides exhibit similar biological functions[8].Accordingly,we refined the algorithm and constructed an online service of GPS 1.1,which can predict p-sites for 71 PK clusters[9].Later,we presented GPS 2.0 and 2.1(renamed as Group-based Prediction System),in which two methods matrix mutation(MaM)and motif length selection(MLS)were designed to improve the prediction accuracy,whereas the scoring strategy was not changed[10,11].Using 3417 known PK-specific p-sites as a training data set,GPS 2.1 contains 213 individual predictors,and can hierarchically predict specific p-sites for 408 human PKs[11].We also developed GPS 2.2,3.0,and 4.0 algorithms,which are used for the prediction of post-translational modification(PTM)sites other than p-sites[16-18].In particular,it should be noted that other bioinformaticians also put great efforts into the prediction of kinase-specific p-sites.At least 36 computational programs have been developed(Table S1).
In this study,we collected 15,194 experimentally identified PK-specific p-sites as the training data set.Updated from the GPS 2.1 algorithm,we replaced the MLS method by developing a new approach named position weight determination(PWD).PWD uses the logistic regression(LR)[19]to rapidly determine position-specific weight values of flanking sequences around psites.The LR algorithm is also used to modify the MaM method into scoring matrix optimization(SMO)for improving the accuracy of prediction.The leave-one-out(LOO)validation and n-fold cross-validations were conducted to evaluate the performance of GPS 5.0,which shows a highly competitive accuracy in comparison with other existing tools.In GPS 5.0,we separately constructed 466 and 93 individual predictors to computationally analyze phosphoserine (pS)/phosphothreonine(pT)and phosphotyrosine(pY)residues specifically modified by serine/threonine kinases and tyrosine kinases,respectively.Since a number of serine/threonine and tyrosine kinases also modify pY and pS/pT sites,respectively,we further constructed 58 additional predictors for these dual-specificity PKs.In GPS 5.0,we developed two modules including the classical module and the species-specific module.In the classical module,we constructed 617 single predictors for computationally identifying specific p-sites of 479 human PKs.The species-specific module can predict p-sites of 44,795 PKs in 161 eukaryotes.We anticipate GPS 5.0 can help to generate high-confidence candidates for the discovery of new phosphorylation events.
Method
During the past decade,the GPS algorithm has been continuously maintained and improved [8-11]. Our fundamental hypothesis is that similar short peptides bear similar biochemical properties for the modification.Thus,we defined a phosphorylation site peptide PSP(m,n)as a pS,pT,or pY amino acid flanked by m residues upstream and n residues downstream.Then we used an amino acid substitution matrix,e.g.,BLOSUM62,to calculate the similarity between two PSP(m,n)peptides.This basic scoring strategy has been reserved in all versions of GPS algorithms,although GPS 2.1 implemented two methods,MLS and MaM,for performance improvement[11].For each PK cluster,MLS determines an optimal motif length around p-sites since different PKs recognize distinct motifs with different lengths,whereas MaM generates an optimal matrix for better estimating PSP(m,n)similarity.Since different positions around p-sites might contribute differentially to the phosphorylation specificity,GPS 2.2 added a method of weight training(WT)to determine a weight value for each position after the MLS manipulation[16].To process large data sets,we added a k-means clustering procedure in GPS 3.0 to cluster PTM sites into multiple groups[17],whereas GPS 4.0 adopted a particle swarm optimization(PSO)to rapidly determine parameters in the steps of MLS,MaM,and/or WT[18].
Here,we hypothesized that long flanking regions around psites might be generally and differentially important for the recognition of PKs,which are bulky molecules to interact with phosphorylatable residues.Thus,the weight value at each position rather than the motif length could be directly and rapidly optimized by the LR algorithm[19].Because the numbers of psites for most PK clusters are lower than 1000(Tables S2 and S3),the k-means clustering is not necessary.In this regard,GPS 5.0 was updated from GPS 2.1,and comprises two parts,including the scoring strategy and performance improvement.
In the step of the scoring strategy,the average similarity score(S)between a PSP(30,30)peptide P and peptides around all known p-sites in the training data set is defined as:
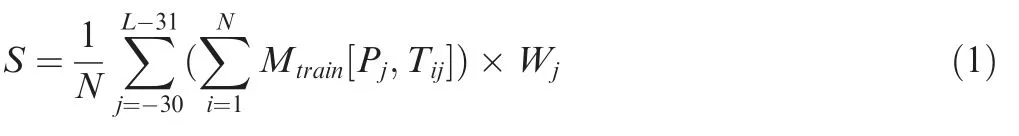
where L is the length of the PSP(30,30)peptide(L=61 representing a relatively long flanking region).N is the number of known p-sites in the positive data set.Tijis the amino acid at position j around a known p-site Ti(i=1,2,3,...,N).Wjis the weight value of position j,and Mtraindenotes the optimized amino acid substitution matrix in this study.
The performance improvement procedure comprises two steps,and we updated MLS and MaM into PWD and SMO,respectively.
PWD
We first used the amino acid substitution matrix BLOSUM62(MBLOSUM62)to calculate an average similarity score at the position j of a PSP(30,30)peptide P as

Initially,the weight value of each position Wjin the PSP(30,30)peptide was set to 1.Then we used the one-vs-rest(OVR)classifier with the ridge(L2)penalty of the LR algorithm to optimize Wjvalues,by using the‘‘newton-cg”solver in the class LogisticRegressionCV of scikit-learn v0.21.0 (https://scikitlearn.org/),an extensively used machine learning(ML)toolbox[19].To avoid over-fitting,such a procedure was repeated for 100 times and 10-fold cross-validation was conducted to determine the inverse of regularization strength at each time.Receiver operating characteristic(ROC)curves were illustrated,and area under curve(AUC)values were calculated.The optimal Wjvectors were determined based on the highest AUC value:
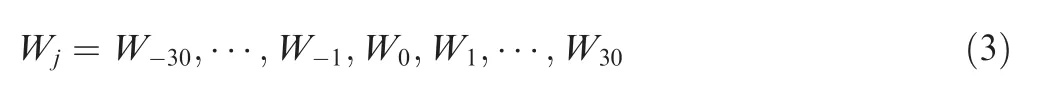
In order to evaluate position-specific contributions of flanking regions around p-sites for different PK clusters,the Wjvectors were normalized into-1 to 1 based on the maximum absolute value.
SMO
The average similarity score of an amino acid a in the given PSP(30,30)peptide P and a residue b in peptides around all known p-sites is defined as Sab:
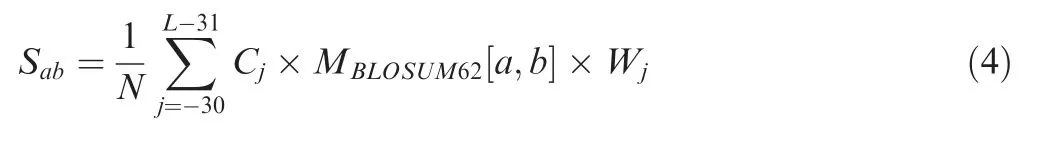
where Cjis the number of ab amino acid pairs at position j.In BLOSUM62,there are 24 types of characters including 20 types of amino acids and 4 non-canonical characters(B,aspartic acid or asparagine;Z,glutamic acid or glutamine;X,any one type of 20 amino acids;*,the ending of protein sequence).Thus,a number of[24×(24+1)]/2=300 unique Sabscores(Sab=Sba)were generated.Then,the same LR algorithm was used to optimize all of Sabscores to produce a new matrix
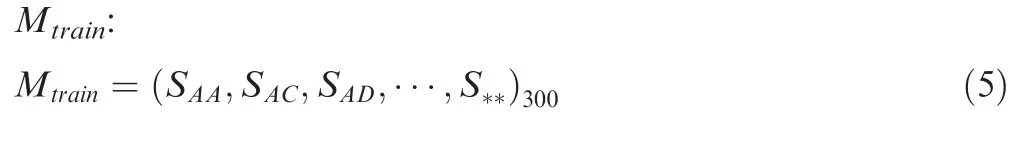
Implementation
First,we took 3417 experimentally identified p-sites used in GPS 2.1[11],and further conducted a literature curation to collect 10,225 site-specific kinase-substrate relations(ssKSRs).Also,we obtained 12,031 known PK-specific p-sites from the file‘‘Kinase_Substrate_Dataset.gz”(Last modified on May 02,2019)of PhosphoSitePlus(https://www.phosphosite.org/),a widely used phosphorylation database[20].In total,our benchmark data set contained 23,195 ssKSRs for 15,194 unique p-sites(Figure 1 and Table S4).
As previously described[10],we downloaded the hierarchical classifications of human PKs at various levels(group,family,subfamily,and single PK)from Kinase.com/KinBase(http://kinase.com/web/current/kinbase/genes/SpeciesID/9606/),thus far the best annotated resource for PKs[21].Due to the fact that multiple aliases are present for each human PK,here we only used the standard gene names taken from iEKPD(http://iekpd.biocuckoo.org),which adopted the classification rationales of Kinase.com/KinBase to characterize and classify eukaryotic PKs at group and family levels[22].Based on the regulatory PK information,we classified known PK-specific p-sites into different PK clusters at group,family,subfamily,and single PK levels.The PK clusters with <3 p-sites were not further considered.It is well known that serine/threonine and tyrosine kinases usually modify pS/pT and pY sites,respectively.However,we found that a considerable number of serine/threonine and tyrosine kinases could additionally phosphorylate pY and pS/pT sites with important functions,respectively. For example, interferon-induced, doublestranded RNA-activated protein kinase(EIF2AK2/PKR)in the Other/PEK family is a typical serine/threonine PK that phosphorylates a human tumor suppressor p53 on S392 through physical interaction to regulate gene expression[23].EIF2AK2/PKR also exhibits tyrosine kinase activity and modifies human cyclin-dependent kinase 1(CDK1)at Y4 to promote its ubiquitination and proteasomal degradation[24].Moreover, human proto-oncogene tyrosine-protein kinase receptor RET in the tyrosine kinase(TK)/Ret family regulates the tyrosine kinase activity of focal adhesion kinase(FAK)through phosphorylating its Y576 and Y577[25].Human RET also modifies an important stress-responsive activating transcription factor 4 (ATF4) at four threonine residues,including T107,T114,T115,and T119,to inhibit ATF4-mediated apoptosis[26].Thus,we added a class of‘‘Dual”for the prediction of atypical p-sites of these dual-specificity PKs(Figure 1).
Then,the GPS 5.0 algorithm was adopted to individually train a computational model for each PK cluster.In the classical module,we totally constructed 617 single predictors for computationally identifying specific p-sites of 479 human PKs, including 58 predictors for the dual-specificity PKs(Figure 1).For the development of the species-specific module,eukaryotic PKs pre-classified at group and family levels were taken from iEKPD[22].For each eukaryotic organism,PKs were reserved if their corresponding predictors at the family level could be obtained.In total,the species-specific module could predict the PK-specific p-sites of 44,795 PKs in 161 species(Figure 1).
As previously described[10],we randomly generated 10,000 PSP(30,30)peptides based on the frequencies of the 20 amino acid residues in the training data set to estimate the false positive rate(FPR)of predictions.For each PK cluster,the process was repeated 20 times, and the average value was determined as the final FPR.The high,medium,and low thresholds were adopted with FPRs of 2%,6%,and 10%,respectively,for serine/threonine kinases.Likewise,FPRs of 4%,9%,and 15%were adopted for high,medium,and low thresholds for tyrosine kinases. The online service of the classical module of GPS 5.0 was implemented in PHP and JavaScript.We also integrated two web servers,IUPred[27]and NetSurfP[28],to predict surface accessibilities,disorder regions,and secondary structures of inputted proteins.The stand-alone packages of GPS 5.0 were developed in JAVA for supporting three major operation systems including Windows,Linux,and Mac OS(Figure 1).

Figure 2 Performance evaluation of GPS 5.0
Performance evaluation and comparison
As previously described [10], four standard measurements including accuracy(Ac),sensitivity(Sn),specificity(Sp),and Matthews correlation coefficient (MCC) were adopted to evaluate the performance and robustness of the GPS 5.0.The self-consistency validations were calculated for all PK clusters(Tables S2 and S3).Also,10-fold cross-validations were performed for 245 PK categories wit ≥30 p-sites(Table S2),and LOO validations were conducted for other PK categories(Table S3).The high congruence of different validation results indicated the promising accuracy and robustness of GPS 5.0(Tables S2 and S3).
To further demonstrate the superiority of GPS 5.0,we compared the prediction performance of GPS 5.0 with that of other existing predictors,such as GPS 2.1[11],ScanSite 4.0[12],NetPhos3.1[14],KinasePhos 1.0[15],KinasePhos 2.0[29],and MusiteDeep[13](Figure 2).Due to the page limitation,four typical PK families including CDK,casein kinase 2 (CK2), protein kinase A (PKA), and mitogen-activated protein kinase (MAPK) were selected for demonstration(Figure 2).For each PK family,we directly submitted its corresponding training data set into each tool to calculate the performance and compared with the 10-fold cross-validation result of GPS 5.0.The ROC curves of GPS 2.1[11]and MusiteDeep[13]were illustrated,while the Sn and 1-Sp values of ScanSite 4.0[12]were calculated at high,medium,low,and minimum thresholds separately.The default cut-off scores of NetPhos3.1[14]and KinasePhos 2.0[29]were adopted,whereas Sn values of KinasePhos 1.0[15]with Sp at 100%,95%, and 90% were computed separately. As shown in Figure 2A,we found that GPS 5.0 achieved a highly competitive accuracy with MusiteDeep,a deep learning-based predictor[13].The prediction performance of GPS 5.0 was much better than that of other tools,including GPS 2.1[11](Figure 2A).It should be noted that MusiteDeep only constructed 5 PK-specific predictors at the family level,whereas GPS 5.0 could predict for much more PK families, such as CaM kinase/protein kinase D (CAMK/PKD) and TK/Tec(Figure 2B).The pS/pT and pY sites differentially modified by dual-specificity tyrosine phosphorylation-regulated kinase(DYRK)could also be accurately predicted(Figure 2B).
For the four PK families of CDK,CK2,PKA,and MAPK,we further compared the performance of the two new methods in GPS 5.0 with that of previous approaches implemented in GPS 2.1.For each PK family,the AUC values of MLS,PWD,MaM,and SMO were exclusively calculated from the 10-fold cross-validations.Our results demonstrate that PWD and SMO perform better than MLS and MaM in p-site prediction as indicated by higher AUC values for all four PK families tested(Figure 3A).In addition,three ML algorithms in scikitlearn,including support vector machines(SVMs),random forest(RF),and k-nearest neighbor(KNN),were adopted for training models and compared with GPS 5.0.As shown in Figure 3B,GPS5.0 achieved higher AUC values of the 10-fold cross-validations than other algorithms for all four PK families tested.
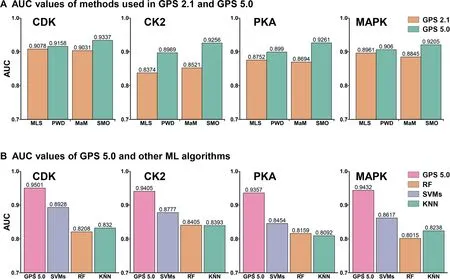
Figure 3 Performance comparison between GPS 5.0,GPS 2.1,and other ML algorithms
Usage of GPS 5.0
For convenience,the stand-alone packages of GPS 5.0 are recommended(Figure 4).The main interface is the classical module of GPS 5.0,which contains three parts,including the hierarchical classification tree of PK categories shown in the left panel(Figure 4A),which enables the selection of PKs at four levels including group,family,sub-family,and single PK.In the lower-right panel,users could provide one or multiple protein sequences in FASTA format and select a threshold(Figure 4B).By left-clicking on the‘Submit’button,the prediction results will be presented in the upper-right panel as a tabular list containing position,code,PK,flanking peptide,score,and cutoff for a predicted p-site(marked in red)(Figure 4C).Alternatively,user could load a demo sequence by clicking the‘Example’button,or clear the inputs by clicking the‘Clear’button(Figure 4).
In GPS 5.0,human Beclin-1,an important autophagyrelated (ATG) protein and tumor suppressor [30,31], was chosen as an example for the prediction of kinase-specific p-sites. It has been reported that the S234 and S295 of Beclin-1 are phosphorylated by Akt,which inhibits autophagy by regulating the interaction between Beclin-1 and 14-3-3 proteins[30].The predictions of GPS 5.0 are highly consistent with experimental results.Two additional p-sites,S10 and S90,were predicted under a medium threshold.Whether the two p-sites are really phosphorylated by Akt remains to be experimentally validated.
Also,GPS 5.0 web server was developed in a user-friendly manner(Figure 5A,Figure S1).For each PK predictor,a sequence logo is illustrated by the R package ggseqlogo[32]with the PSP(30,30)items of its positive data set,and a simplified logo icon is added for each prediction result(Figure S1A).A column entitled‘‘Source”was added to denote whether a potential ssKSR was previously reported by the literature(Exp.)or just a prediction(Pred.)(Figure S1A).Besides the presentation and statistics of the predicted results,structural features such as secondary structures,surface accessibilities,and disorder regions could also be predicted and shown by IUPred and NetSurfP(Figure S1).In IUPred,the disorder propensity values range from 0 to 1,and an amino acid residue with a calculated score>0.5 would be considered as disordered[27].In NetSurfP,the relative surface area(RSA)was calculated for measuring the surface accessibility,and an amino acid with an RSA value>0.25 would be taken as an exposed residue[28].From protein sequences,NetSurfP could also predict three types of potential secondary structures,including α-helix,β-strand,and coil,for each amino acid residue[28].

Figure 4 Interface of the classical module in GPS 5.0 software package
To further exhibit the superiority of GPS 5.0,other known PKs that phosphorylate human Beclin-1 were collected from the literature[31,33-36].Unc-51-like kinase 1(ULK1)has been reported to induce autophagy through phosphorylating Beclin-1 at S15[33],while S90 is phosphorylated by CAMK2 to promote the ubiquitination of Beclin-1 for the activation of autophagy[34].Also,it is known that two pY sites(Y229 and Y233) in Beclin-1 are phosphorylated by epidermal growth factor receptor(EGFR),which is primarily responsible for the suppression of autophagy[31].The prediction results of GPS 5.0 covered most of the known kinase-specific p-sites.Moreover,two additional p-sites,S64 and S177 of Beclin-1,were predicted by GPS5.0 to be specifically modified by ULK1(Figures 5B and S1).
Future developments
In this study,we updated a highly useful tool named GPS 5.0 for the prediction of PK-specific p-sites,including a classical module(Figure 4)and a species-specific module(Figure S2).In the former,there were 617 individual predictors constructed for predicting p-sites of 479 human PKs,whereas kinasespecific p-sites of 44,795 PKs could be predicted for 161 eukaryotes in the latter.In GPS 5.0,two novel methods,PWD and SMO,were developed to improve the training efficiency and performance of the previous developed GPS 2.1 algorithm[11].
For each PK predictor,the information content Riwas calculated in bits for the position i in the alignment as previously described[37]:

Figure 5 Prediction of kinase-specific p-sites in human Beclin-1
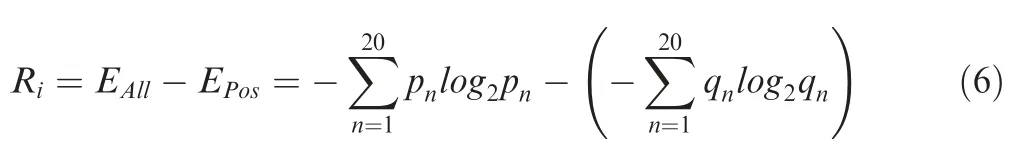
where EAlland EPosindicate Shannon entropies measured from the PSP(30,30)items in all phosphorylated proteins and in the PK-specific positive data set,respectively.The symbol n denotes one of the 20 types of amino acid residues,whereas pnand qnindicate the observed frequencies of n estimated from the background and foreground data sets,respectively.The Rivalues were separately calculated for serine/threonine PKs and tyrosine PKs,while the middle p-sites were not included for the computation.Then,Pearson correlation coefficient(PCC)values are pairwisely calculated between the Riscores and the outputs of PWD training processes for 617 individual PK predictors(Table S5).The average PCC value was 0.606,which was increased to 0.656 if only PK predictors with ≥30 p-sites were considered.Several instances for the correlation between the information content and the PWD output were shown(Figure S3).For example,weight values for AGC/Akt at positions-5 and-3 were determined as 0.8454 and 1.0000. Such a result follows the canonical motif R-X-R-X-X-S/T of the Akt family[38](Figure S3).Also,only the position+1 for Atypical/PIKK/ATM was determined as 1.0000, which is consistent with the S/T-Q motif of ATM/ATR[39].Moreover,the weight values of 0.5677 and 1.0000 at positions -2 and +1 are consistent with the P-X-S/T-P motif of CMGC/MAPK[40],and the weight value of 1.0000 at the+3 position supports a known motif Y-X-X-P of TK/Abl[41](Table S5 and Figure S3).In this regard,higher position weights derived from PWD are generally consistent with information contents of known PK consensus motifs.
Since December 2004,the online service of GPS 1.1 and local packages of GPS 2.0 as well as 2.1 have been freely accessible to academic use for nearly 15 years[9-11].In future,GPS 5.0 will be continuously maintained and improved.The computational models will be updated if new experimentally characterized kinase-specific p-sites are available.In addition,we are currently testing various types of methods including both traditional ML algorithms and deep-learning algorithms,which will hopefully further improve the accuracy of GPS.It is also worth mentioning that only sequence information has been considered at the current stage,and we will test structural features and further integrate both sequence and structural features to improve the performance.We believe that GPS 5.0 could serve as a high-profile tool and provide useful information for further studies of phosphorylation.
Availability
The online service and local packages of GPS 5.0 were freely available for academic use at http://gps.biocuckoo.cn.Speciesspecific predictions are available either from the web server at http://gps.biocuckoo.cn/online_species.php,or in GPS 5.0 software packages by clicking‘Tools’in the menu bar.The benchmark data set for training was provided in Table S4.
Authors’contributions
YX conceived,designed,and supervised the study.CW and HX collected the data,designed the algorithm,performed the analysis,as well as constructed the online service and local packages.SL,WD,JZ,YZ,YS,and DP contributed to data analysis.YX,CW,and HX wrote the manuscript.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
Funding for open access charge:Special Project on Precision Medicine under the National Key R&D Program of China(Grant Nos. 2017YFC0906600 and 2018YFC0910500),National Natural Science Foundation of China(Grant Nos.31671360,81701567,and 31801095),National Program for Support of Top-Notch Young Professionals, Changjiang Scholars Program of China.This study is also supported by the program for HUST Academic Frontier Youth Team,Fundamental Research Funds for the Central Universities,China(Grant Nos.2017KFXKJC001 and 2019kfyRCPY043),and China Postdoctoral Science Foundation (Grant Nos.2018M642816 and 2018M632870).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2020.01.001.
ORCID
0000-0002-0920-8639(Wang C)
0000-0003-2086-3893(Xu H)
0000-0002-1177-5480(Lin S)
0000-0002-5052-9151(Deng W)
0000-0002-9285-8914(Zhou J)
0000-0002-2018-7728(Zhang Y)
0000-0002-8509-3503(Shi Y)
0000-0002-1249-3741(Peng D)
0000-0002-9403-6869(Xue Y)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- The Elements of Data Sharing
- The Birth of Bio-data Science:Trends,Expectations,and Applications
- Role of Long Non-coding RNAs in Reprogramming to Induced Pluripotency
- CRISPR Screens Identify Essential Cell Growth Mediators in BRAF Inhibitor-resistant Melanoma
- Epitranscriptomic 5-Methylcytosine Profile in PM2.5-induced Mouse Pulmonary Fibrosis
- Procleave:Predicting Protease-specific Substrate Cleavage Sites by Combining Sequence and Structural Information