Huntington病家系分析与临床研究
2020-07-27曾倩曹中伟孙俊卿
曾倩 曹中伟 孙俊卿

[摘要] 亨廷頓病(HD)是一种典型的常染色体显性遗传的神经变性疾病,该病是由单基因HTT突变导致的遗传性疾病,IT15基因中胞嘧啶-腺嘌呤-鸟嘌呤(CAG)n重复拷贝的异常扩展是家系发生HD的发病基础。重复序列>36次时发病,HD患者出现变异蛋白的累积和神经元死亡病理特征,伴随着行为、认知、精神方面的障碍。本文收集2017~2019年于内蒙古自治区人民医院神经内科就诊HD患者及部分家系成员的详细临床资料,探讨亨廷顿病家系中遗传规律和临床特征,绘制完整的家谱图,并对患者进行IT15基因CAG三核苷酸重复次数的检测,用于该病的基因诊断及症状前诊断。在两家系中确诊3例患者,致病CAG重复拷贝数≥40次。家系分析中发现,父系遗传有遗传早现现象。晚期HD患者存在较典型的临床表现,但早期症状多样且不典型,临床诊断困难,行为学改变,影像学检查有重要的参考价值,基因诊断可确诊该疾病。
[关键词] 亨廷顿病;家系;临床研究;基因诊断;重复序列
[中图分类号] R742.2 [文献标识码] A [文章编号] 1673-7210(2020)06(b)-0087-04
Family analysis and clinical research of Huntington disease
ZENG Qian1 CAO Zhongwei2 SUN Junqing3
1.Department of Neurology, Inner Mongolia People′s Hospital, Inner Mongolia Autonomous Region, Hohhot 010017, China; 2.Department of Thyroid Breast Surgery, Inner Mongolia People′s Hospital, Inner Mongolia Autonomous Region, Hohhot 010017, China; 3.Department of Medical Clinic, Inner Mongolia People′s Hospital, Inner Mongolia Autonomous Region, Hohhot 010017, China
[Abstract] Huntington′s disease (HD) is a typical autosomal dominant neurodegenerative disease. The disease is a genetic disease caused by a single gene HTT mutation. In the IT15 gene, cytosine-adenine-guanine (CAG) n repeats of the abnormal expansion of copies is the basis of HD in the family. The disease occurs, when the repeat sequence>36 times. HD patients have accumulated variant protein and neuronal death pathological features, accompanied by behavioral, cognitive, and mental disorders. This article collects detailed clinical data of HD patients and some family members treated in the Department of Neurology of Inner Mongolia People′s Hospital from 2017 to 2019, discusses the genetic rules and clinical characteristics of HD families, draws a complete family tree, and tests the number of CAG trinucleotide repeats of IT15 gene in patients, using for genetic diagnosis and pre-symptomatic diagnosis of the disease. Three patients were diagnosed in both lines, and the causative CAG repeat copy number was≥40 times. In the analysis of the family, it was found that there was a premature phenomenon of paternal inheritance. The patients with advanced HD have more typical clinical manifestations, but the early symptoms are diverse and atypical, the clinical diagnosis is difficult, the behavior changes, and the imaging examination have important reference value. Genetic diagnosis can confirm the disease.
[Key words] Huntington′s disease; Pedigree; Clinical research; Genetic diagnosis; Repeat sequences
享廷顿病(Huntington′s disease,HD)是一种常染色体显性遗传性疾病。临床特征为进行性加重舞蹈样不自主运动、精神异常和痴呆“三联症”,病变为纹状体投射性γ氨基丁酸(GABA)神经元和大脑皮质运动区锥体细胞变性死亡。多于中年(35~40岁)起病,有家族史,偶有散发。男女均可发病,起病隐匿,慢性进行性加重,患病后平均生存15年。IT15为致病基因,定位于4p16.3,由胞嘧啶-腺嘌呤-鸟嘌呤(CAG)重复扩增变异产生Huntingtin蛋白而致病。现将内蒙古自治区人民医院神经内科两个HD家系中14例患者临床资料和遗传特征分析如下:
1 资料与方法
1.1 一般资料
两家系中有2例为临床诊断明确HD患者,A家系先证者为女性,30岁;B家系先证者为女性,26岁。2例患者分别来自2017年3月、2019年2月内蒙古自治区人民医院神经内科门诊。两家系中共有5名成员接受基因分析,其中男1名,女4名。基因检测阳性者3例,已发病2例;最小年龄3岁,平均14.8岁。
A家系先证者,女,自24岁始逐渐出现语言含糊不清,全身不自主活动,行走不稳,智力减退6年,于2017年3月就诊。6年来,四肢躯干出现不随意活动,尤以双下肢为著,走路不稳,记忆力减退。神经系统检查:神志清,智能减退,伴有严重焦虑、抑郁、易激惹,语言含糊不清,有时嬉笑,口中时有不自主的“吭吭”声音,仪表不整。颅神经无异常,时有伸舌,如努嘴,挤眉弄眼及耸肩动作,四肢有规则舞蹈动作,步态蹒跚,肌张力降低,腱反射减弱,病理征未引出。MRI示尾状核及壳核萎缩,脑室扩大。其父亲有相似病史。
B家系先证者,女,26岁。因言语欠清、行走不稳、四肢不自主动作3余年就诊,主要表现为双侧上肢不自主动作,行走时踩棉花感,行走不稳伴智力下降,精细动作欠灵活,易跌倒。神经系统检查:神志清楚,构音障碍,表情淡漠,抑郁、偶有幻觉,四肢及头颈部不自主舞蹈样动作,四肢肌力5-级、肌张力低,腱反射减弱,共济运动失调阳性,闭目难立征阳性,病理征未引出。头部MRI提示尾状核轻度萎缩。脑电图双侧导联可见中量低波幅θ波及δ波。
1.2 家系调查
据先证者线索追溯家庭成员,A家系其家系血缘关系能记忆的四代23名成员中患本病9例,B家系四代20名成员中患本病5例。见图1。
A家系四代23名成员中有9例发病占39.13%,男女比为4∶5,Ⅱ3和Ⅱ4小家系8名成员中4例发病。本家系发病年龄24~28岁,其发病年龄逐渐出现早现,发病年龄提前。患者于40岁内病故。仅其先证者Ⅲ5及其妹Ⅲ7、Ⅲ9,其女儿Ⅳ3存活。Ⅰ1本人患病,其3子(Ⅱ1、Ⅱ3、Ⅱ5)发病。Ⅱ1本人及1女(Ⅲ1)发病。Ⅱ3本人、2女(Ⅲ5、Ⅲ7)及孙女(Ⅳ3)发病。Ⅱ5本人及1女(Ⅲ9)发病。Ⅲ5为先证者。见图1A。
B家系四代20名成员中患本病5例,发病占25%,男女比为1∶4,Ⅱ4和Ⅱ6小家系11名成员中3例发病。本家系发病年龄23~32岁,其发病年龄逐渐出现早现,发病年龄提前。患者于38岁内病故。Ⅰ1本人患病,其1子(Ⅱ3)、2女(Ⅱ4、Ⅱ6)发病。Ⅱ4本人及1女(Ⅲ2)发病。目前先证者Ⅲ2存活。见图1B。
2 影像学检查和基因检测
A家系中Ⅱ3和Ⅱ4小家系7名成员肝功能肾功能检查正常。对其先证者Ⅲ5,其妹Ⅲ7,女儿Ⅳ3进行头部MRI检查,发现Ⅲ5、Ⅲ7、Ⅳ3 MRI示尾状核及壳核萎缩,脑室扩大。B家系Ⅲ2小家系3名成员进行血生化、脑电图、头颅核磁检查,先证者Ⅲ2 MRI示基底节区轻度脑萎缩,脑电图双侧导联可见中量低波幅θ波及δ波。
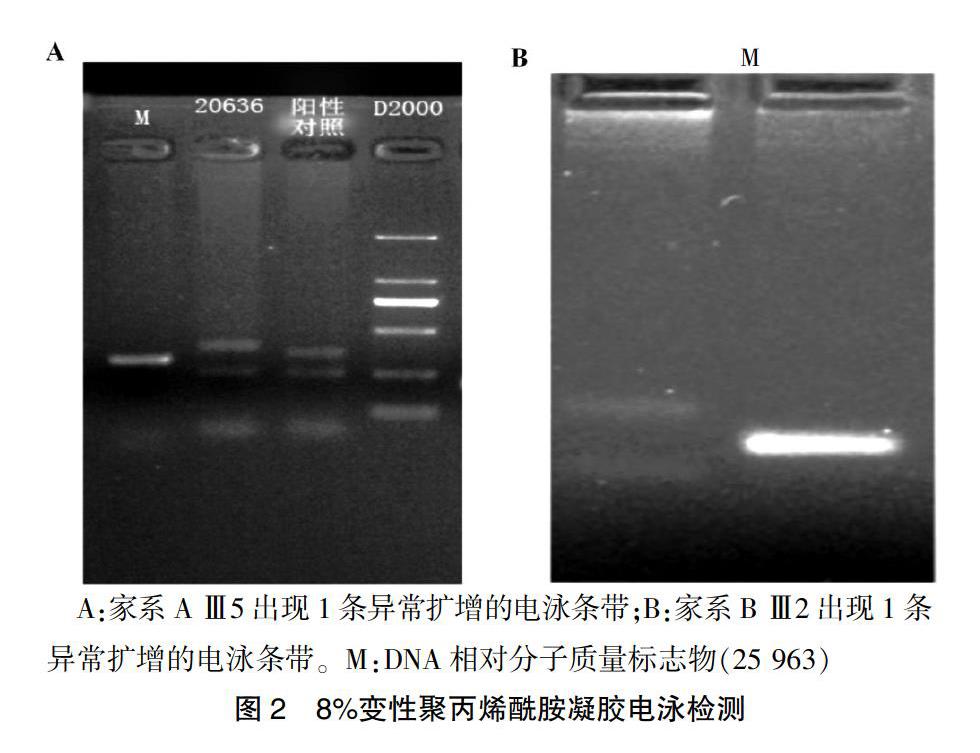
IT15基因的(CAG)n重复拷贝检测,经患者及家属签署知情同意书后,对A家系先证者Ⅲ5及其女儿Ⅳ3;B家系Ⅲ2及其女儿Ⅳ1、儿子Ⅳ2采集外周血各10 mL送福建省神经病学研究所进行基因检测。在目的基因三核苷酸重复序列的两端侧翼序列设计引物,以受试者外周血白细胞基因组DNA为模板并应用聚合酶链式反应(PCR)。对患者重复序列进行扩增,PCR扩增产物经煮沸变性后由8%变性聚丙烯酰胺凝胶电泳(PAGE)分离大小片段,经固定、银染及显色后摄像并进行结果分析。8%变性聚丙烯酰胺凝胶电泳检测显示异常电泳条带,结果在患者1条染色体上IT15基因1号外显子(CAG)n重复次数≥40次,结合临床症状提示A家系母女为HD患者,B家系先证者为HD患者。见图2。
3 讨论
HD又称Huntington舞蹈病,慢性进行性舞蹈病,于1872年由美国医生George Huntington详细报道而得名,临床特征为慢性进行性加重的舞蹈样动作,精神异常和痴呆。中国/日本HD患病率为0.4/10万,而欧洲/北美以及大洋洲患病率为5.7/10万[1]。该病目前缺少有效的治疗方法,且HD症状多种多样,常被误诊,如老年性舞蹈病、小舞蹈症、肝豆状核变性、亚急性硬化性全脑炎后震颤麻痹、脊髓小脑共济失调(SCA)和肌张力障碍等疾病的有类似症状[2],便给HD的准确临床诊断增加了难度。因此,寻找早期准确诊断HD或病程发展评估HD的生物学标志物至关重要[3]。
两家系患者起病于成人23~32歲;主要临床表现为舞蹈样动作,精神异常,智力减退;病程10~15年呈慢性进行性发展;影像学显示患者尾状核,壳核萎缩,脑室扩大;IT15基因(CAG)n重复拷贝检测结果在患者1条染色体上IT15基因1号外显示的(CAG)n重复次数≥40次。两家系14例患者可明确诊断HD。
两家系患者发病年龄,病情进展及病情严重程度不一这些与文献的报道相一致。除三联征外,HD还可有共济运动失调,吞咽困难和构音障碍与国内外报道相似[4]。目前报道的HD症状前患者及患者中多伴有焦虑及抑郁,且发生率较高,可发生在病程的任何阶段,有10%~80%患者伴有不同程度的抑郁风险[5-6],伴有焦虑的发生率为13%~71%[5,7-9]。关于焦虑抑郁与病情发展的关系研究,至今未达成统一结论[9-12]。出现HD症状前到真正发病有比较长时间的潜伏期,故基因检测阳性结果给患者及家属带来一定的心理影响。在接受基因测试时部分患者的焦虑抑郁水平最高[13-14],因为检测结果可能让家系成员中的年轻人面临婚育和遗传风险、承担家庭负担及社会歧视等问题[15]。因此,对家系中成员有必要进行心理及遗传方面的咨询、随访。
我国几个地区发现44个家系共333例患者,多数起病缓慢隐匿,待症状明显后才进行就诊,延误了病情。自该病的遗传基础为IT15基因内(CAG)n三核苷酸异常扩展性重复于1993年被明确后,通过PAG技术鉴定(CAG)n重复序列成为对HD进行分子水平诊断的最简便、最直接的途径[16]。
A家系由父亲遗传子代中发病年龄一代早于一代,其Ⅱ3和Ⅱ4小家系中Ⅱ3发病为28岁,而子代Ⅲ5、Ⅲ7发病为24岁,子代发病年龄明显早于父代,且存在性别差异,这一性别差异可能与精子形成的减数分裂过程中DNA的不稳定性密切相关[17]。发病年龄受多种因素影响,如遗传背景、父系遗传中父亲年龄,其他修饰基因、环境因素及随机过程也可影响发病的准确年龄[18]。也有研究证实[19],HD患者发病年龄与CAG重复次数存在负相关,即CAG重复次数越多,发病年龄将会越早,进一步证实HD患者遗传早现规律。
在近年有研究认为,CAG当重复拷贝数达到一定值(>50次)时,发病年龄与(CAG)n重复次数存在明显的相关性[20]。而当重复拷贝数为40~50次时,二者相关性不明显,CAG重复数>50次可作为HD早发的预测“阈值”[21]。关于CAG重复数与疾病严重程度的关系存在争议,大多数学者倾向于重复数愈多疾病愈严重[21-22]。有研究显示[23],CAG重复较低的患者,神经系统症状进展较缓慢,认知和功能障碍较轻。
目前HD尚缺乏有效的治疗方法,均为对症治疗。如针对舞蹈症状可选择用氟哌啶醇等,但需慎重,特别注意锥体外系的不良反应。一些实验性治疗方案如Caspase抑制剂,线粒体功能保护剂,Htt聚合体形成抑制剂等;国外有在临床尝试用金刚烷胺、百尤解、利鲁唑等药物治疗HD,对部分患者的运动和精神症状有一定效果。
由于HD为常染色体显性遗传性疾病,外显完全,且多在成年期发病,子代中有50%再现风险,危害严重。对高危家族成员进行筛查,发现HD基因突变者,进行开展产前诊断,无创的产前诊断[24]能为亨廷顿病家系阻断HD致病基因在家系中传递,从根源上避免患儿出生。
[参考文献]
[1] Tamara P,Katie W,Lundy D,et al. The incidence and prevalence of Huntington′s disease:a systematic review and meta-analysis [J]. Mov Disord,2012,27(9):1083-1091.
[2] Nicholas TP,Elaine BS,Thomas WP. Technical standards and guidelines for Huntington disease testing [J]. Genet Med,2004,6(1):61-65.
[3] Jean-Marc B,馮璐扬.亨廷顿舞蹈病:教学型综述[J].中国神经精神疾病杂志,2015,41(10):577-591.
[4] 柯国秀,刘春风,林芳,等.Hungington病的临床和遗传特征[J].临床神经病学杂志,2008,21(1):15-17.
[5] van Duijn E,Kingma EM,van der Mast RC. Psychopathology in verified Huntington′s disease gene carriers [J]. Neuropsychiatry Clin Neurosci,2007,19(4):441-448.
[6] Eric AE,Ji-In K,David C,et al. Longitudinal Psy-chiatric Symptoms in Prodromal Huntington′s Disease:A Decade of Data [J]. Am J Psychiatry,2016,173(2):184-192.
[7] Sarah JT,Douglas RL,Blair RL,et al. Biological and clinical manifestations of Huntington′s disease in the longi-tudinal TRACK-HD study:cross-sectional analysis of baseline data [J]. Lancet Neurol,2009,8(9):791-801.
[8] Dale M,Van Duijn E. Anxiety in Huntington′s Disease [J]. Neuropsychiatry Clin Neurosci,2015,27(4):262-271.
[9] Dale M,Maltby J,Shimozaki S,et al. Disease stage,but not sex,predicts depression and psychological distress in Huntington′s disease:A European population study [J]. Psychosom Res,2016,80:17-22.
[10] Thompson JC,Harrs J,Sollom AC,et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington′s disease [J]. Neuropsychiatry Clin Neurosci,2012,24(1):53-60.
[11] Kingma EM,van Duijn E,Timman R,et al. Behavioural problems in Huntington′s disease using the Problem Behaviours Assessment [J]. Gen Hosp Psychiatry,2008,30(2):155-161.
[12] Paulsen JS,Nehl C,Hoth KF,et al. Depression and stages of Huntington′s disease [J]. Neuropsychiatry Clin Neurosci,2005,17(4):496-502.
[13] Downing N,Smith MM,Beglinger LJ,et al. Perceivedstress in prodromal Huntington disease [J]. Psychol Health,2012,27(2):196-209.
[14] Paulsen JS,Nance M,Kim JI,et al. A review of quality of life after predictive testing for and earlier identification of neurodegenerative diseases [J]. Prog Neurobiol,2013,110:2-28.
[15] Ellison M. The impact of Huntington disease on young people [J]. Handb Clin Neurol,2017,144:179-182.
[16] 林芳,陈志欣,刘春风,等.亨廷顿舞蹈病—家系四例[J].中华医学遗传杂志,2006,23:486.
[17] 曹广娜,包新华,卢红梅,等.2个Huntington病家系临床特征及CAG重复性分析[J].北京大学学报:医学版,2011,43(2):163-167.
[18] Souitieri F,Cannella M,Gial lonardo P,et al. Onset and preonset studies to define the Huntington′s disease natutal history [J]. Brain Res Bull 2001,56(3/4):233-238.
[19] 邢世会,陈玲,陈曦,等.Huntington舞蹈病4个家系IT15基因突变的研究[J].中国神经精神疾病杂志,2013,10(39):592-596.
[20] Jiang H,Sun YM,Hao Y,et al. Huntingtin gene CAG repeatnumbers in Chinese patients with Huntington′s disease and controls [J]. Eur J Neurol,2014,21(4):637-642.
[21] Rosenblatt A,Liang KY,zhou H,et al. The association of CAG repeat length with Clinical progression in Huntington disease [J]. Neuology,2006,66(7):1016-1020.
[22] Aziz NA,Jurgens CK,Landwehmeyer GB,et al. Nomal and mutant HTT interact to affect clinical severity and progression in Huntington disease 9 [J]. Neurology,2009, 73(16):1280-1285.
[23] Ravina B,Roner M,Constantinesu R,et al. The relationship betwen CAG Repeat length and clinical progression in Huntington′s disease [J]. MoV Disord,2008,23(9):1223-1227.
[24] Jessica ME van den Oever,Emilia KB,Ilse F,et al. Noninvasive prenatal diagnosis of Huntington disease:detection of the paternally inherited expanded CAG repeat in maternal plasma [J]. Prenat Diagn,2015,35(10):945-949.
(收稿日期:2020-01-03 本文編辑:王晓晔)
