二代测序技术在烟草中的应用进展
2020-07-15苏代发杨俊誉刘志华崔晓龙
何 沛,苏代发,杨俊誉,肖 炜,刘志华*,崔晓龙*
(1.云南中烟工业有限责任公司技术中心,中国云南昆明650231;2.云南大学生命科学学院,中国云南昆明650091)
核苷酸序列测定作为最重要的分子生物学分析方法之一,不仅能为遗传信息的揭示和基因表达调控等基础生物学研究提供重要数据,而且在基因诊断和基因治疗等应用研究中也发挥着重要的作用。为了进行快速、准确的基因测序,Sanger等[1]于1977年提出了快速测定DNA序列的链终止法,此后的四十多年时间里,测序技术取得了相当大的发展,出现了新的测序技术(包括二代测序技术等)[2]。然而,随着遗传信息的不断揭示,第一代测序技术已经不能满足科学研究的需要,因此2005年第二代测序技术(next-generation sequencing,NGS)应运而生[3]。第二代测序技术克服了第一代测序技术的缺点,凸显出其快速、高通量和低成本的巨大优势,这一细致、全面的测序方法为基因组和转录组的深入研究提供了可能[4]。
与Sanger测序技术相比,NGS是一种一次能对几十万到几百万的核酸分子进行测序的高通量测序技术,这种高通量测序使得人们对一个物种的转录组和基因组进行细致、全貌的分析成为可能。目前,NGS在生物学中的应用包括:全基因组测序,主要目的在于测得某一物种的全部基因组,如拟南芥[5]、大肠杆菌[6]等;宏基因组测序,其主要目的在于解读微生物群体的多样性与丰度,探求微生物与环境、微生物与宿主之间的关系,发掘和研究新的、具有特定功能的基因,目前广泛应用于海洋生物、植物、人类疾病等候选病毒资源的发掘及病毒多样性的研究[7~8];转录组测序,涵盖全部mRNA和small RNA的测序与分析[8];扩增子测序,包括16S rDNA测序、18S rDNA测序、转录间隔区(internal transcribed spacer,ITS)测序及目标区域扩增子测序等,是一种高靶向性的测序方法,用于分析特定基因组区域中的基因变异。
在市场上占据优势的三大主要测序平台包括Roche公司的454平台、Illumina公司的Solexa和HiSeq平台及Applied Biosystems(ABI)公司的SOLiD平台[9~14]。由于454及SOLiD平台存在序列读长、费用等方面的瑕疵,所以当前二代测序主要以Illumina为测序平台。本文对烟草中所涉及的二代测序技术进行了综述,以期为相关领域研究者提供新的思路。
1 7种烟草属植物的基因组测序
第一个烟草基因组测序项目于2003年由美国烟草基因组计划启动[15~16],对栽培种烟草Hicks Broadleaf的开放阅读框进行了测序[17],并于2007年完成测序工作,得到了约689 Mb的序列。Matsuoka等[18]为了解析植物细胞在细胞分裂过程中和细胞分裂终止后基因表达的变化,用cDNA基因芯片研究了烟草BY-2细胞系生长过程中基因表达的变化,得到了约9 200条表达序列标签(expression sequence tags,ESTs)片段,与之对应的基因有7 000个,研究发现:对数期细胞主要表达DNA/染色体复制基因的同源物;而平稳期(stationary phase)细胞则表达多种与受体激酶、信号转导和转录因子相关的基因。法国与英国合作发起了欧洲烟草EST计划[19],其以烤烟品种K326、白肋烟21和白肋烟TN86为材料,选取植株生长的不同阶段(发芽期、幼苗期、打顶前后期和成熟期)分别制备植物样本(种子、根、茎、梗、叶片和花)。该项研究从由56 000份样本组成的11个标准化cDNA文库中得到了一个庞大的烟草EST数据集,其研究结果为推动烟草基因研究进程做出了重大贡献。
中国于2010年启动烟草基因组计划,2011年完成了绒毛状烟草(Nicotiana tomentosiformis)和林烟草(Nicotiana sylvestris)的全基因组序列图谱[20];2012—2016年完成了标记数最多、密度最高的烟草分子遗传连锁图谱[21~24];2014年构建了首张烟草单倍体型图,揭示了烟草百年育种进程[20];同时,建立了烟草基因组数据库,其汇集基因序列、基因芯片、突变体、代谢组等所有产出的数据资源,已经成为国际上覆盖烟草基因组数据最全面、体系最完整、最有应用和研究价值的数据库和云计算平台[20]。
目前已用二代测序技术对N.tomentosiformis等7种烟草属植物的基因组进行了测序,其基因组大小从1.70 Gb到4.60 Gb不等[25~28],具体信息见表1。
此外,现有研究中也有对烟草叶绿体进行全基因组测序的相关报道。Asaf等[29]对烟草N.otophora的叶绿体全基因组进行了测序,发现其叶绿体基因组有156 073 bp,共有163个基因,蛋白质编码基因有110个;而N.sylvestris、N.tabacum、N.tomentosiformis和N.undulata的叶绿体基因组分别为 155 941 bp、155 943 bp、155 745 bp 和155 863 bp,所含有的基因分别为140个、144个、140个和156个,蛋白质编码基因分别为111个、98个、110个和111个。
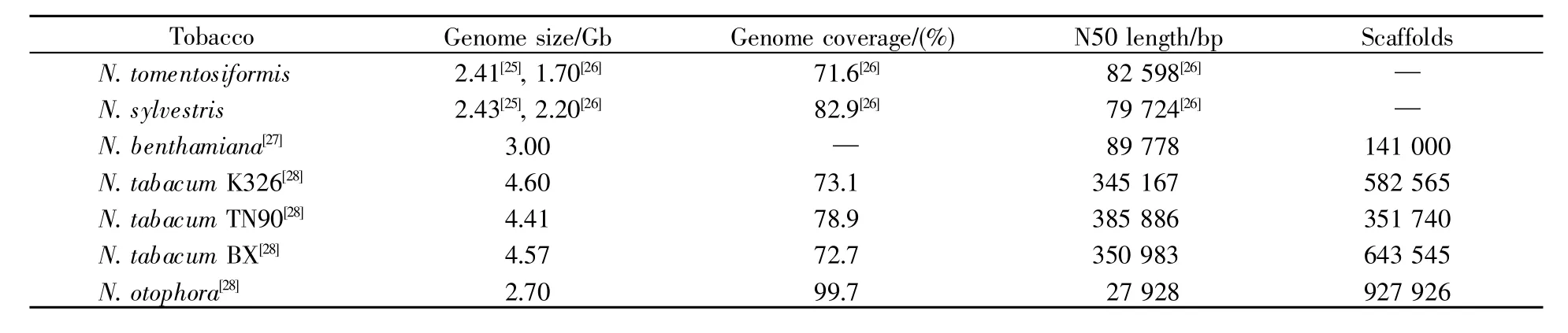
表1 7种烟草属植物的基因组测序Table 1 Genome sequencing of seven tobacco species
2 宏基因组测序
Huang等[30]以烟草K326的烤烟叶为材料,通过免培养方式对陈化与未陈化烤烟叶的细菌多样性进行分析,结果显示从未陈化、陈化的烤烟叶中分别鉴定出23个和15个细菌,芽孢杆菌属(Bacillus)和假单胞菌属(Pseudomonas)是陈化与未陈化烤烟叶中的优势属。Su等[31]以陈化与未陈化津巴布韦烤烟叶为材料,用免培养方式对其中细菌的多样性和系统发育进行了研究,结果显示从陈化、未陈化的烤烟叶中分别得到65个和84个运算分类单元(operational taxonomic units,OTUs),两种烤烟叶中的优势种各有不同;来自两种材料的细菌可以分为两个分支,其中陈化烤烟叶中的细菌又可以分为两个独立的亚分支。Wang等[32~33]以来自红河和楚雄两地的陈化烤烟(aging flue-cured tobaccos,AFTs)为材料,用高通量测序对陈化烤烟表面的细菌多样性进行了研究,同时对细菌潜在的遗传能力进行了预测,结果表明在陈化烤烟表面的主要细菌可以分为7门、36科、48属,芽孢杆菌(Bacillus spp.)在两种陈化烤烟表面普遍存在;该研究团队通过重建未观察到的状态对群落进行系统发育研究(phylogenetic investigation of communities by reconstruction of unobserved states,PICRUSt),发现陈化烤烟微生物群落具有诸如香味的生物合成及降解有害化合物的潜在代谢能力。
Tyx等[34]以美国产的干鼻烟、湿鼻烟和苏丹toombak为材料,用二代测序技术对其中细菌的多样性和分类丰度(taxonomic abundances)进行了研究,结果表明细菌可以分为4门、33科,美产干鼻烟包括4门,湿鼻烟的优势门为硬壁菌门(Firmicutes),toombak的优势门为放线菌门(Actinobacteria)和硬壁菌门(Firmicutes),这为进一步研究无烟烟草产品独特的微生物和化学环境奠定了基础。Al-Hebshi等[35]以来自4个国家的无烟烟草(包括 American moist snuff,Swedish snus,Sudanese toombaks和Yemeni shammah)为材料,通过二代测序技术对其中细菌的组成和功能进行了研究,结果显示:在物种水平上得到491个分类群(specieslevel taxa),分属于11门、178属;瑞典鼻烟的物种丰富度和多样性最高,而Yemeni shammah最低,只含芽孢杆菌(Bacillus spp.);美国鼻烟中芽孢杆菌属(Bacillus)、类芽孢杆菌属(Paenibacillus)和大洋芽胞杆菌(Oceanobacillus spp.)的丰度最高;在假定的“高致癌性”产品中,编码镉/锌和镍转运系统的基因很丰富。
常安然等[36]以四川凉山州健康现蕾期烟草的根际土为材料,利用Illumina MiSeq高通量测序技术对根际土壤细菌进行分析,发现环境因子有机质、pH、含水量和土壤速效氮对健康现蕾期烟草根际土壤细菌有显著影响。曹毅等[37]以贵州省烟草科学研究院福泉烟草青枯病病圃内的土壤为材料,用454焦磷酸测序技术对土壤中的细菌组成进行了分析,其研究结果暗示部分细菌群落可能与烟草青枯病的发生相关。施河丽等[38]通过宏基因组测序对青枯病发病烟株与健康烟株的根际土壤的细菌群落进行了研究,发现健康烟株根际土壤的细菌多样性、pH、养分及有益菌均要高于患病烟株根际土壤。张笑宇等[39]分析了易感黑胫病烟田与健康烟田烟草在不同生育期内的根际土壤微生物动态,发现在烟草生长过程中微生物多样性呈下降趋势,健康烟田的细菌多样性优于患病烟田,烟草患黑胫病主要与病原菌增加、土壤微生物群落结构改变及多样性降低有关。
张艺洁等[40]采用Illumina MiSeq高通量测序技术,对不同施肥处理下连作植烟土壤进行了细菌和真菌的多样性分析。其研究结果表明施肥可以明显改变连作土壤中细菌与真菌的结构,为种烟区土壤环境因子与微生物群落多样性研究提供了理论依据。Xiao等[41]以湖南省境内连续种植烟草1年、4年、5年和12年的土壤为材料,通过Illumina平台对土壤微生物群落进行了分析。其研究结果表明冬季休耕期与作物生长后期的土壤微生物群落存在显著差异;作物的发病与土壤细菌群落、冬季休耕期、作物生长后期某些细菌属的存在显著相关;在冬季休耕期,土壤细菌的丰度低,而由冬季休耕期向作物生长后期转变时,土壤中的细菌丰度增加;在作物生长后期,由于土壤生物量低,故而这个时期倾向于病害高发。Lei等[42]以来自安徽省池州市的土壤为材料,通过高通量测序研究了烟-稻轮作耕作方式对土壤细菌多样性和组成的影响。其研究结果表明用生石灰或白云石粉尘覆盖烟草-水稻秸秆、减少施肥会对土壤性质及微生物多样性和组成产生影响;水稻秸秆还田可显著提高土壤微生物的多样性和丰度,而随着施肥量的增加,土壤微生物的多样性和丰度明显降低,以上信息提示施肥量对细菌群落组成有影响。
3 转录组测序
穆淑媛[7]通过Illumina HiSeqTM2000平台对烟草(N.tabacum)NC89进行了转录组测序,在对所得序列进行注释后发现约有14.61%的单基因簇(unigenes)与植物的生长发育相关。龙妮[2]以N.glauca、N.noctiflora、N.cordifolia、N.knightiana、N.setchellii和N.tomentosiformis 6种烟草野生种为材料,通过Illumina HiSeq 2000平台首次对这6种野生烟草的转录组进行了报道,并对其中的抗性基因、与抗性相关的转录因子及烟碱转导基因进行了鉴定,为烟草抗病品种的选育提供了理论依据。表2列举了N.sylvestris[26]、N.tomentosiformis[26]和N.benthamiana[43]3种烟属植物基因转录组测序的相关信息,其测序所用材料涉及根、叶、花等烟草组织。
此外,Faino等[44]通过深度RNA测序测定了感染大丽轮枝菌(Verticillium dahliae)烟草N.benthamiana的转录组,发现其总转录组大小为38.7 Mb,与拟南芥的转录组相当。其研究结果为病原胁迫下的茄科模式植物提供了一个转录本目录(catalogue of transcripts)。段胜常[45]以接种长柄链格孢菌(Alternaria longipes)和链格孢菌(Alternaria alternata)后的烟草V2和NC89为材料,通过Illumina HiSeq 2000平台对其RNA进行转录组测序,筛选出了大量潜在的抗病基因,为烟草疾病预防及烟草育种方面的研究奠定了基础。王新[46]通过对接种青枯菌菌株后的易感烟草和抗病烟草进行转录组测序,筛选出了大量以上调为主的差异基因,并发现在接种青枯菌后,烟草会启动相关基因的表达及多条代谢通路以抵御病原菌的侵染。
Fan等[47]以感染甜菜坏死黄脉病毒(beet necrotic yellow vein virus,BNYVV)的烟草N.benthamiana为材料,通过基于转录组的深度测序揭示了烟草对含有或缺失RNA4的BNYVV侵染的应答。结果表明由甜菜坏死黄脉病毒的RNA4所引起的涉及RNA沉默、泛素-蛋白酶体途径、纤维素合成和赤霉素代谢的基因的表达改变可在植物上表现出严重的症状。Geng等[48]以烟草N.benthamiana为材料,通过转录组测序研究了烟草脉带花叶病毒(tobacco vein banding mosaic virus,TVBMV)野生型及减毒突变体接种对烟草转录组的影响,结果表明接种野生型及突变体1 d后,与翻译相关的基因表达上调,而与脂质合成和代谢、应对胞外及外部刺激相关的基因则被下调,在接种10 d后,与光合作用相关的基因被抑制;野生型及突变体对参与RNA沉默途径的基因有不同程度的干扰;感染野生型病毒后,水杨酸和乙烯信号通路被诱导,而茉莉酸信号通路却被抑制。Huang等[49]以烟草N.tabacum为材料,通过二代测序技术对感染番茄带状斑原卵病毒(tomato zonate spot orthotospovirus,TZSV)的烟草进行了转录组分析,在对烟草转录组从头组装后鉴定出135 395个单基因簇(unigenes);烟草感染TZSV后得到2 102个差异表达基因(differentially expressed genes,DEGs),其中1 518个DEGs被诱导,而584个DEGs则被抑制,该研究结果有助于进一步了解植物对正痘病毒(orthotospovirus)感染的复杂反应机制。Li等[50]通过HiSeqTM2500平台对感染烟草曲茎病毒(tobacco curly shoot virus,TbCSV)的烟草N.benthamiana进行了转录组测序,发现油菜素内酯(brassinosteroid,BR)和茉莉酸(jasmonic acid,JA)的合成及转导相关基因显著发生了改变,当植物(烟草植株)受到病毒侵染时,与植物防御系统相关的基因显著上调表达,这些结果为烟草抵御病原菌侵扰奠定了研究基础。
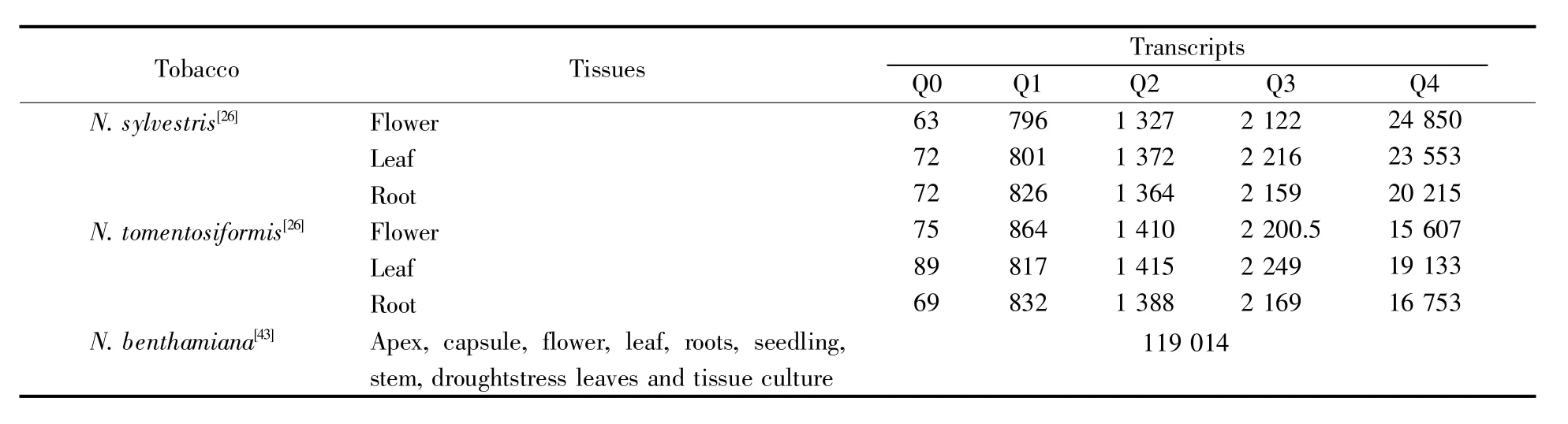
表2 3种烟属植物的基因转录组测序Table 2 Transcriptome sequencing of three tobacco species

表3 二代测序技术用于烟草病毒的鉴定Table 3 Tobacco virus detection using next-generation sequencing technology
Chen等[51]以烟草系K326为材料,通过对烟草叶片进行干旱胁迫和干旱后浇水研究了烟草叶片中的干旱胁迫相关基因及microRNA,功能注释表明表达显著上调的基因与抗氧化、刺激和应激反应相关,而与细胞周期和光合作用过程相关的基因则表达下调;在干旱条件下,5个microRNA家族(miR398、miR390、miR162、miR166 和miR168)存在差异调节,该研究结果为从多个分子遗传水平上进一步研究烟草响应干旱胁迫的分子机制奠定了基础。张柳等[52]借助二代测序技术研究了烟草在逆境胁迫下(低降雨量)的基因表达情况,结果表明雨量降低时上调表达的基因大多是与抗旱以及离子运输或者次生代谢物合成有关的基因(包括干旱胁迫基因及与黄酮类物质和萜类吲哚生物碱合成相关的基因)。
4 病毒检测与鉴定
二代测序技术除了可用于基因组测序、宏基因组测序和转录组测序外,还可用于烟草病毒的检测与鉴定。王芳等[53]以安徽省的烟草样品为材料,通过二代测序技术对烟草干扰小RNA(small interfering RNA,siRNA)进行了测序,以检测烟草RNA病毒。结果显示该方法可用于检测烟草中的天然RNA病毒。王浩军等[54]用二代测序技术对安徽皖南烟区的siRNA进行了测序,建立了一种可在田间对烟草病毒病进行检测和发现烟草新病毒的体系,以实现对大田烟草病毒的调查。刘悦等[55]用二代测序技术建立了一种可用于检测植物病毒的方法,为通过宏基因组检测植物病毒奠定了基础。二代测序技术不仅可对已有烟草病毒进行检测,还可对某些地区新发现的烟草病毒进行鉴定。当前已有将二代测序技术用于烟草病毒鉴定的相关报道[56~57],表3所列病毒中的BrYV-AH和ChiVMV为安徽烟区首次发现。
5 展望
虽然二代测序技术在当前市场上占有主导地位,但仍面临仪器昂贵等劣势,已经无法满足部分生命科学的研究。随着科技的不断进步、测序技术的不断发展,以单分子实时测序技术为代表的三代测序技术(third-generation sequencing)已应用于酵母[58]、兔[59]、高粱[60]、玉米[61]、小麦[62]和棉花[63]等物种,而在烟草中只有渐狭叶烟草(N.attenuata)和欧布特斯烟草(N.obtusifolia)两个野生烟草进行了第三代测序[11],其他烟草基因组则使用的是第二代测序技术和组装方法,且部分注释工作当时尚未完成。新技术的涌现,将为烟草基因组、转录组等测序工作带来契机,尚未完成基因组与转录组测序的烟草可把握时机,填补烟草基因组测序空缺,扩充并完善烟草基因组与转录组数据库,构建烟草基因组图谱;同时可对之前已完成测序的烟草进行重测序、重注释,以期发现或发掘新基因,便于加快烟草的育种进程及烟草抗逆机制的解析,为烟草行业的蓬勃发展奠定理论依据。
