质谱技术在植物蛋白质组学研究中的应用
2020-07-14闫昕
摘要:植物蛋白质组是植物生理特征的动态反映,在植物生长发育、组织器官分化、抵御外界干扰等生理病理过程中发挥着重要作用。综述了基于质谱技术的植物不同器官、不同发育阶段、响应生物及非生物胁迫过程中的蛋白质组学研究进展,以期对后续植物蛋白质组学的研究提供思路设计和试验方法上的参考。
关键词:植物蛋白质组;质谱;生长发育;胁迫响应
中图分类号:Q94
文献标识码:A
文章编号:0439-8114( 2020) 08-0005-06
D01:10.14088/j.cnki.issn0439-8114.2020.08.001
开放科学(资源服务)标识码(OSID):
鉴定蛋白质的结构和功能已经成为植物科学的重要研究内容。对蛋白质氨基酸序列的研究在蛋白质、多肽与其编码基因研究之间搭建了桥梁,更是研究植物蛋白质生理功能及其基因调控间关系的重要纽带。对蛋白质的研究,使研究者可以更好地了解植物细胞内复杂的调控网络与作用机制,更全面地认识不同的生物学过程。近年来,随着质谱技术(Mass Spectrometry,MS)的不断发展,质谱检测的灵敏度和准确度不断提高,质谱法逐渐取代Edman降解等传统方法,成为蛋白质组学研究的主要手段。
1 基于质谱技术的蛋白质组学概念及常用手段
蛋白质作为生物体不同生物学功能的物质承担者,其结构和生物学特性一直以来都是生物领域的热点问题。1996年,Wilkins等[1]提出并使用蛋白质组学( Proteomics) 一词,它是“protein”及“genome”2个词的结合,指的是对某一特定阶段的某种细胞或者组织器官中所有蛋白质进行检测与研究,包括其表达特征、分布特征、相互作用特征、翻译后修饰特征等。人类含有20 235个基因,对应超过100 000个蛋白质,可见蛋白质组学研究具有高度复杂性及较大的动态范围。不同蛋白质间的数量、分子量及亲疏水性上的差别,加大了蛋白质组学的研究难度[2]。蛋白质组学是表征基因功能最好的方式,基因表达水平的波动可以被蛋白质组学动态地反映出来。
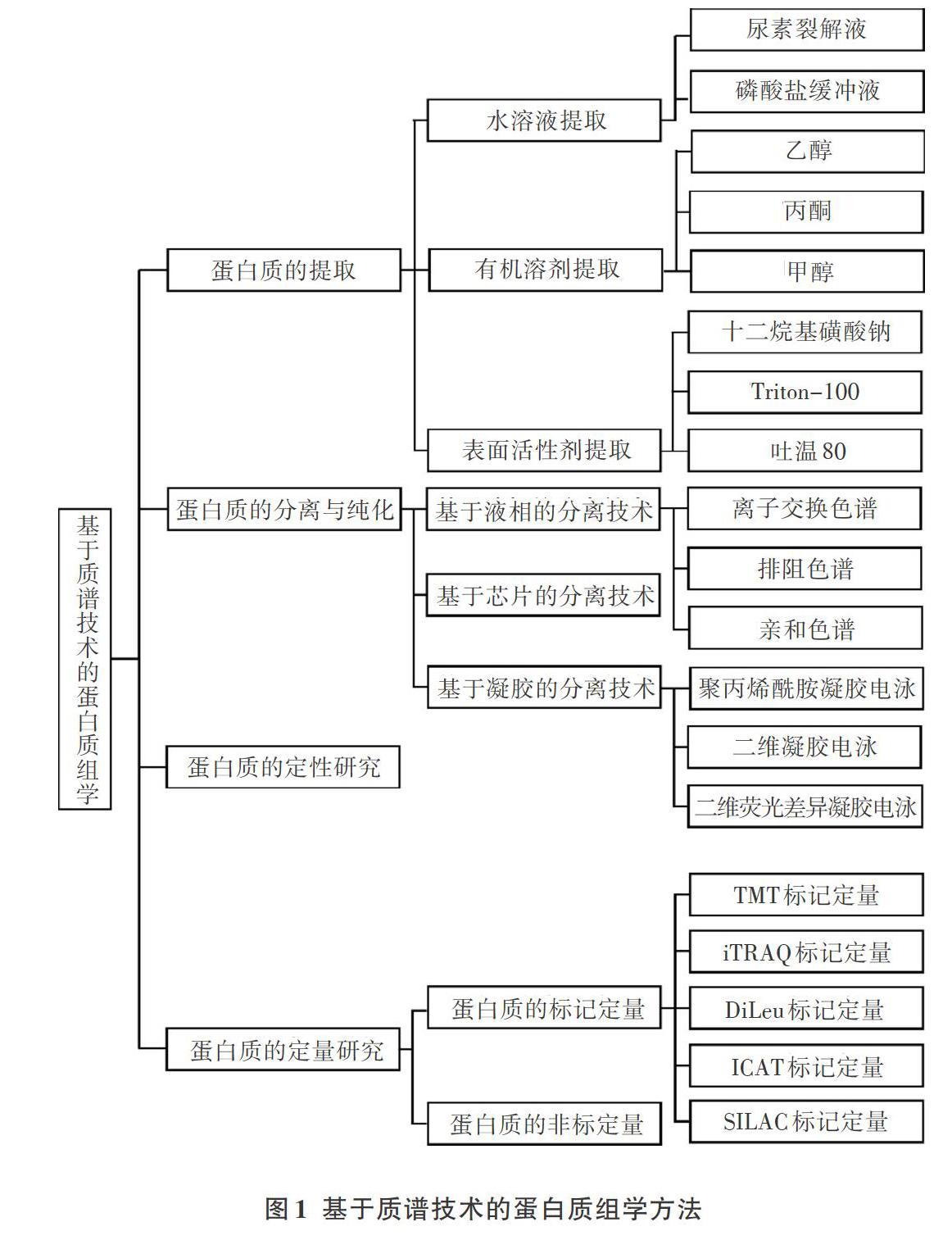
在植物蛋白质组学研究中最常用的策略是自下而上质谱鉴定方法的鸟枪法研究策略。自下而上的研究策略将蛋白质样品进行酶切,酶切后的肽段进行质谱分析,通过将鉴定到的碎片离子进行拼接得到的序列信息与蛋白质数据库的理论酶切结果进行比较,确定待测蛋白质样品的氨基酸序列。鸟枪法的优势在于肽段易于利用色谱等方法进行预分离,分离后的样品易溶解在水溶液中,在高温环境中气化而转化为喷雾状,继而在外加电场的影响下形成带电离子传人质谱,在较为简单的碎裂条件下形成碎片离子而被质谱仪检测到。鸟枪法利用肽段的定性、定量信息来反映蛋白质的结构及表达量,基于这种思路,多种定量分析方法也被开发出来,例如ICAT[3]、TMT[4]、iTRAQ[5]、DiLeu[6]等,如图1所示。
在利用鸟枪法进行蛋白质质谱检测前,常用一定的方法对复杂体系的蛋白质样品进行预分离,以达到更好的鉴定效果。预分离方法主要分为2类,基于液相的蛋白质分离与纯化技术和基于凝胶电泳的分离与纯化技术。基于液相的蛋白质分离与纯化主要是利用不同蛋白质分子的物理、化学性质的不同而实现彼此间的分离,如离子交换色谱( Ion Ex-change Chromatography,IEC)[7]、排阻色谱(Size Ex-clusion Chromatography.SEC)[8]、亲和色谱法(Affini-ty Chromatography, AC)[9]等。
基于凝胶的预分离方法主要是依据蛋白质分子量的不同或者分子量及等电点的不同,将蛋白质在凝胶中分離开来,主要包括十二烷基磺酸钠一聚丙烯酰胺凝胶电泳(Sodium Dodecvl Sulfate-Polyacryl-amide Gel Electrophoresis,SDS-PAGE)[10]、双向凝胶电泳(Two-dimensional Gel Electrophoresis, 2D-Gel)[11]和二维荧光差异凝胶电泳(Two-dimensional Differ-entialGel Electrophoresis,2D-DIGE)[12]。基于凝胶的分离方法,尤其是2D-Gel作为一项成熟的技术已经在蛋白质组学领域应用超过30年。
2 植物组织或细胞器的蛋白质组学分析
2.1 不同器官的蛋白质组学分析
根、叶和果实等作为植物体的重要器官,其蛋白质组得到了广泛的研究。植物的根系在植物生物量中占有很大比重,在植物的水及营养吸收、植物在土壤中的固定及植物对地下环境的感知中发挥着重要作用。Petricka等[13]利用蛋白质组学方法,分离出6种拟南芥根细胞,包括根毛细胞、非根毛表皮细胞、皮质细胞、内胚层细胞、脉管系统细胞和小柱细胞,并对这6种细胞的蛋白质组进行了质谱检测。对鉴定的蛋白质进行分析,发现有超过1/3的蛋白质特异性地在单一类型细胞中表达,其中238个蛋白质仅在根毛中表达,其功能与细胞尖端生长、肽基一脯氨酸羟基化、糖基转移和辅酶Q的生物合成相关。对根毛及非根毛表皮蛋白质组及转录组进行分析,Lan等[14]在含有pEXP7-GFP报告结构的拟南芥植物根部共鉴定到23 034个基因及2 447个高可信蛋白质,其中129个蛋白质在根毛及非根毛组织中差异表达。同时,Lan等L141发现转录产物与蛋白质的丰度并不总是完全对应的,这表明细胞分化过程受到诸多因素调控,包括转录水平、翻译水平、蛋白质的翻译后修饰等,这些因素所产生的影响并不均等。
Martin等[15]利用宽粒子选择监控数据非依赖型非标定量方法( label-free Wide Selected-Ion Monitor-ing Data-Independent Acquisition, WiSIM-DIA).建立了包含来自2 338个番茄果实蛋白质的11 753条特有肽段库,对角质缺陷2基因型(CUTIN DEFI-CIENT2,CD2)番茄与M82基因野生型番茄进行蛋白质组定量研究。共鉴定到1 140个番茄蛋白质,其中67个蛋白质在2种基因型番茄中存在表达差异,差异表达的蛋白质参与表皮素的生物合成。试验证实,CD2蛋白质影响细胞壁的合成、花青素的合成、植物生长并参与压力应答。类似地,Mata等[16]利用超速离心、凝胶分离、胶内酶解及高pH反相色谱分离等技术,提取并分离番茄表皮全蛋白质,继而利用Thermofisher Scientific Q Exactive质谱仪与AB Sciex Triple-TOF 6600质谱仪对其进行分析,共鉴定到8 588个蛋白质。
叶片组织是绿色植物光合作用的主要场所,受到真菌感染的叶片易发生坏死,而导致严重的作物损失。Zhang等[17]利用iTRAQ标记对交链孢菌感染后的茉莉酸不敏感型番茄叶片及野生型叶片进行定量蛋白质组学分析,共鉴定到10 367个蛋白质,其中有2 670个蛋白质存在差异表达。Aryal等[18]利用排阻色谱法对拟南芥叶片的胞质蛋白质进行了分离,鉴定到上百个以稳定复合体形态存在的蛋白质。
2.2 不同细胞器的蛋白质组学分析
随着植物蛋白质组学技术的不断发展及科学探索的不断深入,对植物的蛋白质组研究已经由细胞水平转入亚细胞水平,多种重要细胞器内的蛋白质分布特点得到了详尽描述。叶绿体在细胞代谢中处于中心地位,不仅为细胞提供能量和代谢中间产物,还在某些辅酶、辅酶因子及脂类的生物合成最终步骤中发挥催化作用[18]。借助SDS-PACE电泳技术,Salvato等[19]将提取的马铃薯块茎线粒体全蛋白质进行分离,分离后的蛋白质条带经胶内酶解后通过LTQ Orbitrap XL进行质谱检测。利用4种搜库软件,在马铃薯块茎线粒体中共鉴定到1 060个非冗余蛋白质,其表达量的动态范围高达1 800倍,广泛参与细胞电子传递链的组成、三羧酸循环过程及蛋白质运输结构的组成。此外,这些蛋白质中超过50%的蛋白质发生了一种以上翻译后修饰,最常见的为氧化修饰。
叶绿体作为植物细胞所特有的能量细胞器,是一系列代谢途径的集合,包含多个蛋白质复合体机器,包括光合系统I和光合系统Ⅱ、细胞色素b6f复合体和ATP合成酶等[20,21]。此外,叶绿体也是必需氨基酸、脂肪酸和维生素合成的场所。因此,叶绿体蛋白质表达特点一直以来都是植物学家关注的热点问题。Lande等[22]利用Percoll梯度过滤法从鹰嘴豆中分离叶绿体,并用丙酮及尿素提取叶绿体蛋白质,SDS-PAGE电泳分离及酶切后进行质谱检测,共鉴定到2 451个蛋白质,包括27个异构体。类似地,利用差异蛋白质组学方法,Uberegui等[23]以光对叶绿体蛋白质的影响为研究对象,检测到几种光合作用及碳代谢相关蛋白质以及胞质mRNA加工相关蛋白质的丰度变化,提示执行蛋白质(Executer pro-teins)在光照条件下参与拟南芥的信号传导,参与调控叶绿体代谢及植物对环境变化的响应。
高尔基体在植物細胞壁基质多糖合成、蛋白质糖基化及囊泡转运过程中发挥着重要作用,Parsons等[24]利用密度梯度离心及表面电荷分离技术,从拟南芥中分离出高尔基体膜,利用质谱定量技术共鉴定出371个高尔基体蛋白质,其中包含参与细胞壁合成的重要蛋白质。
3 植物胁迫相关蛋白质组学分析
3.1 生物胁迫
植物的生物胁迫是指植物在生长发育过程中,由于其固着生长的特点,容易受到一些生物体来源的环境胁迫,如病原体感染(包括真菌、细菌、病毒、线虫)[25]、寄生植物干扰、害虫干扰等,植物通过多种生理、生化、细胞和分子水平的变化来抵抗这些胁迫效力。宿主植物主要通过2种方式来抵御病原体等生物胁迫,即组成型抵抗和诱导型抵抗。植物组成型抵抗主要依赖于形态学特征的改变和次生代谢产物的合成,包括细胞壁成分(如木质素、角质和蜡质)、气孔结构、低分子量抗真菌化合物、可以与真菌几丁质结合的凝集素以及核糖体失活蛋白质等[26]。对宿主植物抵抗真菌感染的研究是目前最为深入的,真菌种类包括镰刀菌(Fusarium)、稻瘟病菌(Pyricularia grisea)、炭疽病病原体(AnthracnosePathogens)、大豆锈病菌(Uromyces Appendiculatus)、甘蓝链格孢(Alternaria Brassicicola)及葡萄孢菌(Botr-ytis Cinerea)等。Asano等[27]将镰孢镰刀菌和禾本科镰刀菌接种到拟南芥花与叶片上,通过比较蛋白质组学的方法并结合MALDI-TOF/TOF仪器的使用,鉴定到与病原体感染发病相关的显著上调蛋白质,其中谷胱甘肽S-转移酶(GSTs)作为上调蛋白质被首次发现。GST蛋白质是防御反应蛋白质超家族的一员,在生物体对病原体的应激反应中发挥生理功能。
炭疽病在世界范围内都有发生,尤其是在热带和亚热带湿热地区,这些地带更适合炭疽病病原菌的生长。Fang等[28]利用二维凝胶电泳技术对感染炭疽病菌24、48和72 h后的草莓叶片蛋白质进行分离,并用MALDI-TOF/TOF 4800质谱仪对样品进行差异蛋白质鉴定,共检测到49个差异蛋白质,广泛参与代谢过程、光合作用、能量产生过程,具备抗氧化活性;同时,在参与代谢途径的蛋白质水平上观察到许多变化,包括卡尔文循环、磷酸戊糖途径及糖酵解途径,这些发现为探究非模式生物中的病原体抵抗机制提供了参考。
大多数植物病原细菌是杆状细菌,包括假单胞菌属、黄单胞菌属、欧文氏菌属、土壤杆菌属、棒状杆菌属和根瘤菌属[26]。Parker等[29]以Rio Grande番茄(RG)的2个基因同源品种,即表达Pto基因而与细菌不相容的RC-PtoR以及缺失Prf3基因而具有易感表型的RC-prf3,进行iTRAQ定量,以探究不同时间点蛋白质组的变化对植物抵抗病原体能力的影响。根瘤菌可以通过与豆科植物共生,形成根瘤并固定空气中的氮气从而为植物体提供营养,在农业生产中具有重要作用。Nguyen等[30]采用iTRAQ试剂盒标记根瘤菌感染及未感染的大豆根毛和脱落根全蛋白质,借助Ni-NTA磁珠进行磷酸化蛋白质富集,磷酸化蛋白质组在LTQ Orbitrap Velos质谱仪中进行分析。在2种大豆根毛及脱落根中共鉴定到1 625个磷酸化肽段上1 659个磷酸化位点,分布在1 126个大豆磷酸化蛋白质上。在根瘤菌感染后的大豆根毛及脱落根中,有来自240个磷酸化蛋白质的273条磷酸化肽段含量发生显著变化,数据揭示了大豆根毛磷酸化蛋白质组的独特特征,描述了在根瘤菌感染反应中,激酶一底物和磷酸酶一底物相互作用所形成的复杂网络。
3.2 非生物胁迫
与生物胁迫不同,非生物胁迫主要是指来自无机环境中的干扰因素,如干旱、炎热、寒冷、营养不足、盐分过多或者土壤中的有毒金属等[31]。干旱、盐分和温度胁迫是影响自然界植物地理分布、限制农业植物生产力和威胁粮食安全的主要环境因素。理解植物对非生物胁迫的感知及适应机制,对于提高农业生产力、维护环境的可持续性至关重要。寒冷对水稻的生长发育影响很大,水稻幼苗在低温条件下出现生长迟缓、叶面卷曲及部分叶片死亡等现象。为了研究水稻对低温胁迫的抵御机制,Ji等[32]利用三氟乙酸/丙酮提取耐寒型水稻Fujisaka 5及寒冷敏感型水稻9311的全蛋白质,全蛋白质经二维凝胶电泳分离后进行MALDI-TOF/TOF质谱检测,鉴定到59个抵御寒冷相关蛋白质并对其功能进行了分类。此外,研究者还发现在寒冷条件刺激下,膜组成成分及结构发生了破坏,代谢显著改变,水稻防御体系激活。
干旱是植物生长抑制和作物减产的主要制约因素,全球气候变化使得科学家对干旱问题的关注程度日益增加[33,34]。在干旱胁迫下,叶片气孔闭合,二氧化碳的固定减少,导致通过卡尔文循环的NADP+再生减少。随着光合系统活性和光合运输能力的变化,更多的电子泄露并与氧气反应,进而导致活性氧的产量增加[35]。活性氧水平过高将会引起脂质过氧化、蛋白质氧化以及酶活降低,核酸发生氧化损伤,最终引起细胞死亡。基于此,Tamburino等[36]以番茄叶绿体为研究对象,构建干旱模型及干旱一恢复模型,通过综合利用荧光差异二维凝胶电泳技术与质谱技术,对叶绿体中参与干旱胁迫抵御过程的蛋白质进行了探究。试验发现干旱胁迫条件下,番茄植物出现发育不良,脯氨酸、脱落酸水平升高,晚期胚胎基因转录水平升高。水分缺失对叶绿体蛋白质影响很大,主要影响能量相关蛋白质。
土壤重金属污染是土壤无机物污染中危害较大的种类之一。重金属污染来源有2种途径,自然途径及人为途径。自然途径主要是指成土母质及成土过程中对土壤重金属含量的影响;人为因素则包括工业、交通、农业等带来的重金属污染。聚集在土壤中的重金属由于无法被微生物分解而被植物吸收,进入植物体内,进而通过饮食进入人体。重金属在人体内以有害浓度蓄积,威胁人类健康。研究重金属胁迫下植物的蛋白质水平变化,对于农业生产及环境保护具有重要指导意义。举例来说,镉是一种非必需重金属,高浓度的镉会引发植物代谢活动紊乱,如呼吸和蒸腾抑制、碳水化合物代谢紊乱等,对植物的生长发育造成影响[37]。此外,镉在人体内的蓄积会导致主要器官的慢性损伤[38]。针对镉污染的严重后果,Xu等[39]利用含不同浓度CdCl2的培养液培养萝卜幼苗,结合定量蛋白质组学方法、强阳离子交换法及实时定量PCR技术,对萝卜根部组织全蛋白质进行了表达差异描述。对这些差异蛋白质的功能注释显示,它们主要参与碳水化合物和能量代谢过程、胁迫防御过程及信号传导途径。实时定量PCR结果很好地证实了蛋白质组数据的准确性和高可信度。同时,一些与碳水化合物代谢、活性氧清除、细胞转运及信号传导相关的候选靶标蛋白质也被鉴定出参与萝卜镉胁迫响应的调控,如乙酰辅酶A乙酰转移酶、酰基辅酶A合成酶、草酰辅酶A脱羧酶和磷脂酶Dal等。Du等[40]利用不同浓度的PbCl2处理龙须菜,提取全蛋白质并进行二维凝胶电泳分离,将存在表达差异的蛋白质切下酶解后进行MALDI-TOF/TOF质谱检测。质谱结果显示,不同处理浓度下蛋白质的表达量变化幅度不同,其中有14个铅胁迫相关蛋白质被检测到,功能分析显示其广泛参与多种细胞功能,包括光合作用、能量和蛋白质代谢、碳水化合物运输和代谢、信号传导及抗氧化。超氧化物歧化酶、过氧化物酶、谷胱甘肽巯基转移酶和热休克蛋白70在铅刺激下表达上调。对其他重金属,如锌[41]、汞[42]等也有详尽的研究。
4 植物发育过程中的蛋白质组学分析
植物的发育伴随着基因与蛋白质表达的改变,不同于基因组,蛋白质组可以实时地反映植物体发育不同阶段的特点。对植物发育过程中蛋白质组的研究,可以更深入地了解植物生长发育的机制及其调控方式,对于农业、林业生产具有重要指导意义。Nambu等[43]对不同发育时间段接种的中慢生型百脉根根瘤菌全蛋白质进行了定量分析,以探究根瘤菌与宿主建立固氮共生关系过程中的代谢调节机制。研究者对537个节结成熟过程中的蛋白质表达量变化进行了质谱量化分析,发现碳和氨基酸代谢存在显著变化。此外,中慢生型百脉根根瘤菌在節结形成初期进入缺氮生长状态,而在中期进入富氮生长状态,并在此阶段吸收了氨。类似地,Marond-edze等[44]对不同发育阶段的枣椰树果实进行了蛋白质组学比较分析,深入描述了在水果成熟过程中的蛋白质丰度变化及发挥关键作用的蛋白质。
蓖麻作为一种重要的生物燃料作物,具有重要的应用价值。Shah等[45]对蓖麻植物的4个发育阶段的全蛋白质进行了质谱分析,分别鉴定到了626、613 .449和356个蛋白质,共计882个非冗余蛋白质,并鉴定到2种蓖麻毒素亚型,表明蓖麻毒素的表达并不局限在种子中,可能发挥防御作用。
5 展望
近年来,随着质谱技术的不断发展,植物蛋白质组学的研究更为深入,尤其是经典的模式生物,如拟南芥、大豆、水稻等,从基因水平到蛋白质水平都得到详尽的描述。然而,对于其他植物的蛋白质组学研究仍然停留在较低水平,如玉米、苜蓿、小麦等,虽然有相关文献对其部分蛋白质组学特征进行报道,但仍然缺失对大部分蛋白质的有效鉴定。此外,植物蛋白质的翻译后修饰,如乙酰化、磷酸化、糖基化、甲基化等在植物的生长发育及抵御胁迫的过程中发挥着重要作用,虽然已经建立起基于质谱技术的植物蛋白质翻译后修饰研究方法,但依然缺少有效的分离、富集手段,以提高修饰蛋白质的定性和定量研究的效率及准确度。尽管如此,质谱技术仍然是植物蛋白质组学研究的有力工具,在后续研究中扮演不可或缺的角色。
參考文献:
[1] WILKINSM R,SANCHEZ J C,COOLEYAA,et al. Progress withproteome projects: Why all proteins expressed by a genome shouldbe identified and how to do it[J].Biotechnology&genetic engineer-ing reviews, 1996. 13: 19-50.
[2] PANDEY A, MANN M.Proteomics to study genes and genomes IJl.Nature, 2000 .405( 6788): 837-846.
[3] SCHMIDT F,DONAHOE S,HACENS K,et al.Complementaryanalvsis of the mycobacterium tuberculosis proteome hy two-dimen-sional electrophoresis and isotope-coded affinity tag technology[J].Molecular&cellular proteomics, 2004,3(1):24-42.
[4] DAYON L, HAINARD A,LICKER V, et al_ Relative quantificationof proteins in human cerebrospinal fluids by MS/MS using 6-plexisobaric tags[J]. Anahtical chemistrV, 2008. 80(8):2921-2931.
[5] ROSS P L,HUANG Y N,MARCHESE J N,et al.Multiplexed pro-tein quantitation in Saccharomyces cerevisiae using amine-reactiveisobaric tagging reagents[J]. Molecular&cellular proteomics,2005,3(12): 1154-1169.
[6] FROST D C,GREER T,LI L.High-resolution enahled 12-plexDiLeu isobaric tags for quantitative proteomics[J].Analvtical chem-istry, 2015 .87(3):1646-1654.
[7] GARCIA-CALVO M,PETERSON E P,RASPER D M,et al.Purifi-cation and catalytic properties of human caspase family members[J].Cell death and differentiation, 1999,6(4):362-369.
[8] ARYAL U K,XIONG Y,MCBRIDE Z,et al.A proteomic strategyfor global analysis of plant protein complexes[J]. The plant cell,2014.26( 10): 3867-3882.
[9] HAGE D S,ANGUIZOLA J A,BI C, et al. Pharmaceutical and bio-medical applications of affinitv chromatography: Recent trends anddevelopments E J] . Journal of pharmaceutical and biomedical analy-sis . 2012 . 69 : 93-105.
[10] KUMAR A. SRIVASTAVA N C . SINGH V P, et al. Electrophoret-ic analysis of indian isolates of Mycoplasma agalactiae and Myco-plasma bovii by SDS-PAC.E and immunoblotting [Jl. Veterinarymedicine international. 2014 . 2014 : 892421.
[11] ISLAM N. LONSDALE M. UPADHYAYA N M. et al. Protein ex-traction from mature rice leaves for two-dimensional gel electro-phoresis and its application in proteome analysis [Jl. Proteomics ,2004.4( 7) : 1903-1908.
[12] MAROUGA R, DAVID S. HAWKINS E. The development of theDIGE system : 2D fluorescence difference gel analysis technolo-gy [J]. Analytical and bioanalytical chemistry. 2005. 382 (3) :669-678.
[13] PETRICKA J J. SCHAUER M A, MEGRAW M, et al. The proteinexpression landscape of the Arabidopsis root[ J] . Proceedings of thenational academy of sciences of the United States of America.2012 , 109(18 ) : 6811-6818.
[14] LAN P, LI W. LIN W D , et al. Mapping gene activity of Arabidopsisroot hairs [ J l. Cenome biology , 2013 , 14( 6) : 67.
[15] MARTIN L B, SHERWOOD R W. NICKLAY J J. et al. Applica-tion of wide selected-ion monitoring data-independent acquisitionto identify tomato fruit proteins regulated by the CUTIN DEFI-CIENT2 transcription factor [J]. Proteomics. 2016, 16 ( 15-16) :2081-2094.
[16] MATA C I, FABRE B .HERTOG M L, et al. In-depth characteriza- tion of the tomato fruit pericarp proteome [Jl. Proteomics. 2017, 1-2 : 1600406.
[17] ZHANG M. KOH J, LIU L. et al. Critical role of COIl-dependentjasmonate pathway in AAL toxin induced PCD in tomato revealed by comparative proteomics[ J] . Scientific reports . 2016 , 6 : 28451.
[18] RAO R S, SALVATO F. THAL B, et al. The proteome of higherplant mitochondria[J l. Mitochondrion . 2017 . 33 : 22-37.
[19] SALVATO F. HAVELUND J F. CHEN M. et al. The potato tuhermitochondrial proteome [Jl. Plant physiology. 2014, 164 (2) :637-653.
[20] ZIMORSKI V. KU C . MARTIN W F. et al. Endosymbiotic theoryfor organelle origins [Jl. Current opinion in microbiology, 2014,22 : 38-48.
[21] YADAV K N. SEMCHONOK D A. NOSEK L. et al. Supercomplex-es of plant photosystem I with cytochrome b6f, light-harvestingcomplex II and NDH [ J] . Biochimica et biophysica acta bioenerget-ics.2017 . 1858( I ) : 12-20.
[22] LANDE N V. SUBBA P. BARUA P. et al. Dissecting the chloro-plast proteome of chickpea (Cicer arietirzum L.) provides new in-sights into classical and non-classical functions [ J] . Journal of pro-teomics .2017, 165 : 11-20.
[23] UBEREGUI E . HALL M. LORENZ0 0. et al. An Arabidopsis solu-ble chloroplast proteomic analysis reveals the participation of theexecuter pathway in response to increased light conditions EJl.Journal of experimental botany. 2015 . 66( 7) : 2067-2077.
[24] PARSONS H T. CHRISTIANSEN K. KNIERIM B. et al. Isolationand proteomic characterization of the ArabidopsiJ C.olgi defines functional and novel components involved in plant cell wall biosyn-thesis[J ] .Plant physiology , 2012 . 159( 1 ) : 12-26.
[25] ZHOU L, CHEN F. PAN H . et al. Identifying virulence-associatedgenes using transcriptomic and proteomic association analyses ofthe plant parasitic nematode Bursaphelenchus mucronatu.s [J]. In-ternational journal of molecular sciences . 2016 . 17 ( 9) : 1492.
[26] FANC X. CHEN J, DAI L. et al. Proteomic dissection of plant re-sponses to various pathogens [Jl. Proteomics. 2015. 15 (9) :1525-1543.
[27] ASANO T, KIMURA M. NISHIUCHI T. The defense response inArabidopsis thaliana against Fusarium sporotrichioides [J]. Pro-teorue science .2012. 10( 1 ) : 61.
[28] FANC X P. CHEN W Y. XIN Y. et al. Proteomic analysis of straw-berry leaves infected with Colletotrichum fragariae [ J ] . Journal ofproteomics , 2012 , 75 ( 13 ) : 4074-4090.
[29] PARKER J. KOH J.YOO M J, et al. Quantitative proteomics of to-mato defense against Pseudomonas syringae infection [Jl. Pro-teomics, 2013 . 13( 12-13 ) : 1934-1946.
[30] NGUYEN T H. BRECHENMACHER L. ALDRICH J T. et al.Quantitative phosphoproteomic analysis of soybean root hairs inoc- ulated with Bradyrhizobium japonicum [J]. Molecular & cellularproteomics . 2012. 11( 11 ) : 1140-1155.
[31] ZHU J K. Abiotic stress signaling and responses in plants[Jl. Cell,2016 . 167( 2) : 313-324.
[32] JI L, ZHOU P. ZHU Y. et al. Proteomic analysis of rice seedlingsunder cold stress [ J ] . The protein journal, 2017 . 36( 4) : 299-307.
[33] GHATAK A. CHATURVEDI P, NAGLER M. et al. Comprehen-sive tissue-specific proteome analysis of drought stress responsesin Pennisetum glaucu.m (L.) R. Br. (Pearl millet) [J].Journal of proteomics.2016. 143 : 122-135.
[34] PAUL S, GAYEN D. DATTA S K. et al. Dissecting root proteomeof transgenic rice cultivars unravels metabolic alterations and accu-mulation of novel stress responsive proteins under drought stress[Jl.Plant science : An intemational journal of experimental plant biolo-gy,2015.234: 133-143.
[35] BIEHLER K. FOCK H. Evidence for the contrihution of the me-hler-peroxidase reaction in dissipating excess electrons indrought-stressed wheat [Jl. Plant physiology, 1996, 112 (1) : 265-272.
[36] TAMBURINO R. VITALE M. RUCGIERO A, et al. Chloroplastproteome response to drought stress and recovery in tomato (Sola-nu.m lycopersicum L. ) [Jl. BMC plant biology , 2017. 17( 1 ) : 40.
[37] DALCORSO C, MANARA A. FURINI A. An overview of heavymetal challenge in plants : From roots to shoots [J] . Metallomics : In-tegrated biometal science . 2013 , 5(9) : 1 117-1132.
[38] D'ALESSANDRO A. TAAMALLI M, GEVI F. et al. Cadmiumstress responses in Brassica juncea : Hints from proteomics and me-tabolomics [J]. Journal of proteome research, 2013. 12 (11) :4979-4997.
[39] XU L.WANG Y.ZHANG F . et al. Dissecting root proteome chang-es reveals new insight into cadmium stress response in radish(Raphan LtS sativt.t.s L. ) [Jl. Plant & cell physiology. 2017 , 58 ( 1 1) :1901-1913.
[40] DU H, LIANG H H. JIANG Y. et al. Proteome responses of Cracilaria lemaneiformis exposed to lead stress [ J ] .Marine pollu-tion bulletin , 2018 , 135 : 311-317.
[41] BARKLA B J. VERA-ESTRELLA R. MIRANDA-VERGARA M C . et al. Quantitative proteomics of heavy metal exposure in Arabi-dopsis thaliana reveals alterations in one-carbon metabolism en-zymes upon exposure to zinc [ J ] . Journal of proteomics. 2014. 111 :128-138.
[42] BAIG M A, AHMAD J. BAGHERI R. et al. Proteomic and eco-physiological responses of soybean ( Clycine max L.) root nodulesto Pb and Hg stress[Jl. BMC plant hiology, 2018 , 18( I ) : 283.
[43] NAMBU M. TATSUKAMI Y. MORISAKA H. et al. Quantitativetime-course proteome analysis of Mesorhizobium loti during nodulematuration[ J ] . Journal of proteomics . 2015 . 125 : 112-120.
[44] MARONDEDZE C. Date fruit proteomics during development andripening stages [J]. Methods in molecular biology. 2017, 1638:381-398.
[45 ] SHAH M. TEIXEIRA F M. SOARES E L. et al. Time-course pro-teome analysis of developing extrafloral nectaries of Ricinus com-munis [ J ] . Proteomics . 2016 , 16(4) : 629-633.
作者簡介:闫昕(1993-),女,河北保定人,在读硕士研究生,研究方向为蛋白质组学,(电话)13752017761(电子信箱)664527679@qq.com。
