高效液相色谱法测定咪唑生产工艺中反应液中咪唑及2种杂质2-甲基咪唑和4-甲基咪唑
2020-07-06王冰玉赵文英曹晓荣项曙光
王冰玉,赵文英,曹晓荣,项曙光,∗
(1.青岛科技大学 化工学院,青岛 266042;2.齐鲁师范学院 化学与化工学院,济南 250200)
咪唑,又称甘噁啉或1,3-二氮杂茂,其化学式为C3H4N2,是一种五元杂环化合物,一般为无色棱形结晶体或微黄色结晶体,具有腐蚀性、刺激性、毒性等特点。咪唑是一种重要的化工中间体,在医药、农药、固化剂[1]等方面都有着广泛的应用,其国外市场需求量每年都以30%的速度递增。在工业生产中,咪唑由甲醛、乙二醛和氨水反应得到,其生产过程中反应液(碱性水溶液)的含水量高达80%以上,且会产生2-甲基咪唑和4-甲基咪唑等主要副产物,这将影响后期精馏提纯和咪唑产品的质量。
国内外关于甲基咪唑、乙基咪唑、硝基咪唑的检测方法报道的较多,包括紫外光谱法[2]、液相色谱法[3-4]、气相色谱法[5]、液相色谱-质谱法[6-10]、离子色谱法[11-12]、毛细管电泳法[13-14]等,但咪唑的检测方法报道较少。文献[15]以己烷磺酸钠为离子对试剂,以甲醇-磷酸二氢钾/磷酸缓冲液为流动相进行梯度洗脱,测定了克霉唑阴道片中微量咪唑;文献[16]先用丹磺酰氯进行了柱前衍生,然后用乙酸乙酯萃取、乙腈定容后,采用C18色谱柱进行色谱分离,用乙腈-水为流动相进行梯度洗脱,用荧光检测器测定了烟用添加剂中微量咪唑的含量。
由于咪唑反应液的含水量高且含有多种杂质,不宜采用气相色谱和紫外光谱的分析方法。高效液相色谱法是20世纪70年代发展起来的一项高效、快速的分离分析技术,适用于含水溶液体系中的常量和微量组分的分析。本工作拟采用高效液相色谱法测定反应液中的咪唑及其杂质,以期建立一种高效、准确、快速的分析方法。
1 试验部分
1.1 仪器与试剂
EClass 3100型高效液相色谱仪,配DAD 3100型二极管阵列检测器和Chromsoft型色谱工作站;AL 204型分析天平;FRQ-1002T 型超声清洗仪。
离子对试剂溶液:称取6 g十二烷基硫酸钠、3 g磷酸二氢钾于2 L 的广口瓶中,加入水1 300 mL进行溶解,用磷酸调节pH 为3.5,超声至无气泡,加入870 mL乙腈,超声至无气泡,待用。
单标准储备溶液:称取咪唑(2-甲基咪唑或4-甲基咪)标准品0.100 0 g,加水溶解并定容至100.0 mL,摇匀,配制成1 000 mg·L-1标准储备溶液。
混合标准溶液系列:移取适量的咪唑、2-甲基咪唑、4-甲基咪唑标准储备溶液,用水稀释,配制成10,20,40,60,80,100 mg·L-1的混合标准溶液系列。
咪唑标准品的纯度为99.5%;2-甲基咪唑标准品的纯度为98%;4-甲基咪唑标准品的纯度为98%;十二烷基硫酸钠、磷酸二氢钾、磷酸均为分析纯;乙腈为一级色谱纯;试验用水为超纯水。
1.2 仪器工作条件
Supersil-ODS-B 色谱柱(250 mm×4.6 mm,5μm),柱温30 ℃;流动相为体积比为40∶60的乙腈-离子对试剂溶液;等度洗脱;流量1.0 mL·min-1;检测波长210 nm;进样量20μL。
1.3 试验方法
称取过滤后的咪唑反应液样品0.050 0 g 于100 mL容量瓶中,加水稀释至刻度,摇匀,在仪器工作条件下测定,以外标法定量。
2 结果与讨论
2.1 分析条件优化结果
按照仪器工作条件考察了离子对试剂溶液中十二烷基硫酸钠的质量浓度分别为10,13,16,19,22,25 mmol·L-1,离子对试剂溶液pH 分别为2.5,3.0,3.5,4.0,等度洗脱中离子对试剂溶液在流动相溶液中的比例按5%的降幅从70%逐级降至30%时对20 mg·L-1混合标准溶液中咪唑的峰形、保留时间及咪唑和2-甲基咪唑、4-甲基咪唑分离情况的影响。结果表明:当十二烷基硫酸钠的浓度为10~25 mmol·L-1时,咪唑的保留时间为7.0~9.1 min,其峰面积随着十二烷基硫酸钠质量浓度的增大而减小,综合考虑,确定离子对试剂溶液中十二烷基硫酸钠的浓度为16 mmol·L-1;当离子对试剂溶液pH 为2.5~4.0时,咪唑的保留时间基本不变,峰面积随着pH 的增大稍有降低,综合考虑,确定离子对试剂溶液的pH 为3.5。当离子对试剂溶液在流动相溶液中的比例为70%时,咪唑及杂质的分离较好,但咪唑的保留时间较长,为30min;随着离子对试剂溶液比例的降低,保留时间逐渐缩短,但咪唑及杂质的分离效果变差,综合考虑,确定离子对试剂溶液在流动相溶液中的比例为60%。在此优化条件下,咪唑的保留时间约为8.5 min,2-甲基咪唑和4-甲基咪唑的保留时间分别为9.3,10.1 min。三者的分离度均大于2.0,达到良好分离,且峰形较好,3种物质混合标准溶液的色谱图见图1。

图1 混合标准溶液的色谱图Fig.1 Chromatogram of the mixed standard solution
2.2 标准曲线
按照仪器工作条件对混合标准溶液系列进行测定,以3种待测物的质量浓度为横坐标,其对应的峰面积为纵坐标绘制标准曲线。结果表明:咪唑、2-甲基咪唑和4-甲基咪唑标准曲线的线性范围均为10~100 mg·L-1,其线性回归方程分别为y=50.77x+222.4,y=86.53x+241.3,y=78.34x+325.0;相关系数分别为0.999 6,0.999 1,0.999 5。
2.3 检出限和测定下限
按照仪器工作条件对0.01 mg·L-1混合标准溶液进行测定,以3倍信噪比计算检出限(3S/N),咪唑、2-甲基咪唑、4-甲基咪唑的检出限分别为0.02,0.02,0.03 mg·L-1。以10倍信噪比计算测定下限(10S/N),咪唑、2-甲基咪唑、4-甲基咪唑的测定下限分别为0.07,0.07,0.10 mg·L-1,换算成样品中3种物质的质量分数计,其测定下限分别为0.035%,0.036%,0.051%,满足咪唑反应液中杂质的检测技术指标要求(单个杂质的测定下限不大于0.5%)。
2.4 精密度试验
按照试验方法对反应液重复测定8次,咪唑的测定结果分别为10.10%,10.15%,10.14%,10.11%,10.18%,10.17%,10.14%,10.16%,计算测定值的相对标准偏差(RSD)为0.27%,表明该方法精密度良好。
2.5 准确度试验
在反应液中添加20,100 mg·L-1等2个浓度水平的咪唑标准溶液进行加标回收试验,每个水平重复5次。结果显示:20 mg·L-1添加水平的咪唑回收率分别为99.7%,101%,99.7%,100%,100%,平均回收率为100%;100 mg·L-1添加水平的咪唑回收率分别为99.0%,98.9%,99.5%,99.3%,99.1%,平均回收率为99.2%,回收试验结果表明其准确度满足常量分析的要求。
2.6 方法比对
对同一反应釜中不同的反应时间所取样品(编号为样品1#~样品9#)分别按照本方法和气相色谱法(实验室自建方法)进行测定。在用本方法测定时,样品1#~样品9#中咪唑的测定结果(质量分数)分别为10.71%,10.55%,10.30%,10.14%,10.18%,10.11%,10.12%,10.13%,10.12%,平均值为10.26%,RSD 为2.1%;反应液中咪唑的质量分数自10.71%逐渐降低至10.12%,这是由于在咪唑合成反应初始阶段,出现返混,反应液中咪唑的含量较高,随着反应的进行,返混程度降低,产物趋于稳定;在用气相色谱法测定时,这9个样品中咪唑的测定结果分别为11.07%,10.94%,10.76%,10.52%,10.58%,10.45%,10.45%,10.49%,10.44%,平均值为10.64%,RSD 为2.2%;本方法和实验室自建方法结果吻合,且两者RSD 差异不大,说明了本方法的可靠性。2-甲基咪唑在样品中有检出,但其含量低于测定下限;4-甲基咪唑在样品中未检出。
样品1的色谱图见图2。
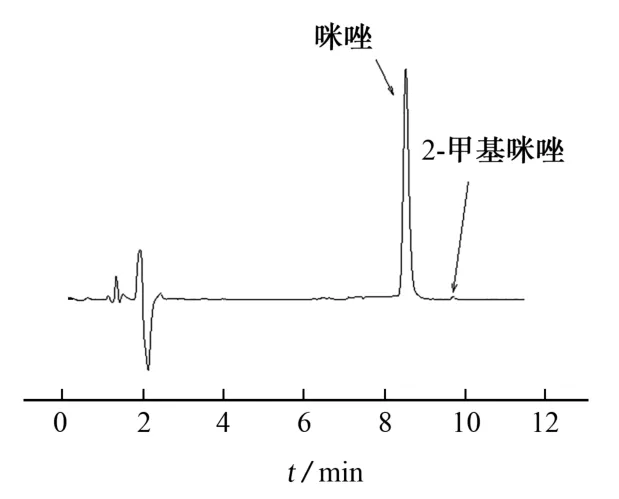
图2 样品1#的色谱图Fig.2 Chromatogram of the sample 1#
本工作建立了高效液相色谱测定反应液中咪唑及2种杂质的方法,该方法简单方便,适用性好,准确性好,效率高,为咪唑合成反应条件的优化、工艺生产过程的监测、产品质量的控制提供可靠、有效的方法。
