先天性肝纤维化伴多囊肾1例报告并文献复习
2020-07-04王亚平杜忠彩刘培牛庆慧梁坤边城
王亚平 杜忠彩 刘培 牛庆慧 梁坤 边城
[摘要] 目的 探讨先天性肝纤维化(CHF)的诊断及治疗。
方法 对我科收治的1例CHF病人的临床资料进行回顾性分析并复习相关文献。
结果 该病人以不明原因肝硬化为首发表现就诊,辗转多家医院,最终通过肝脏穿刺活检病理及基因检测确诊为CHF伴多囊肾。
结论 CHF是一种少见的累及肝脏和肾脏的疾病,临床上主要以门静脉高压和肝功能正常为主要特点,肝脏穿刺活检病理为其诊断金标准,肝移植为最佳根治性方法。
[关键词] 肝硬化;遗传疾病,先天性;多囊肾疾病
[中图分类号] R575.2
[文献标志码] A
[文章编号] 2096-5532(2020)03-0370-03
doi:10.11712/jms.2096-5532.2020.56.047
[开放科学(资源服务)标识码(OSID)]
[网络出版] http://kns.cnki.net/kcms/detail/37.1517.R.20200320.1535.011.html;2020-03-23 15:30:53
CONGENITAL HEPATIC FIBROSIS AND POLYCYSTIC KIDNEY: A CASE REPORT AND LITERATURE REVIEW
WANG Yaping, DU Zhongcai, LIU Pei, NIU Qinghui, LIANG Kun, BIAN Cheng
(Department of Infectious Diseases, The Affiliated Hospital of Qingdao University, Qingdao 266003, China)
[ABSTRACT]ObjectiveTo investigate the diagnosis and treatment of congenital hepatic fibrosis (CHF).
MethodsA retrospective analysis was performed for the clinical data of one patient with CHF who was treated in our department, and related articles were reviewed.
ResultsThe patient attended a hospital due to the initial manifestation of unexplained liver cirrhosis and was examined in several hospitals, and finally he was diagnosed with CHF and polycystic kidney by liver biopsy and gene detection.
ConclusionCHF is a rare disease involving the liver and the kidney, with the main features of portal hypertension and normal li-
ver function. Liver biopsy is the gold standard for diagnosis and liver transplantation is the optimal radical method.
[KEY WORDS]liver cirrhosis; genetic diseases, inborn; polycystic kidney diseases
先天性肝纖维化(CHF)是一种少见的与胆管板畸形(DPM)相关的常染色体隐性遗传性疾病。CHF常合并肾脏疾病[1],且其中一半以上病人合并常染色体隐性遗传性多囊肾(ARPKD),但合并常染色体显性遗传性多囊肾(ADPKD)者少见。CHF病人发病年龄差异较大,但多以青少年为主,临床表现不一,出血风险较高。由于该病临床少见,极容易漏诊、误诊,影响其治疗及预后。本文病人以不明原因肝硬化为首发表现,病程较长,最终通过病理及基因检测确诊为CHF合并ADPKD。现报告如下。
1 病例报告
病人,女,35岁,因“发现脾大、肝硬化10年,肝区疼痛加重1月”于2017-03-14入院。病人10年前因肝区不适于当地医院查体时发现肝硬化,当时上腹部CT示:肝硬化、脾大,脾内低密度,少量腹水;双肾多发囊肿;胆囊结石。胃镜检查示食管静脉曲张。肝脏穿刺活检病理示肝细胞不典型增生,不明原因肝硬化。诊断为“肝硬化、脾大、门静脉高压”,行脾切除术。其后病人多次复查消化系统超声,皆提示脾脏切除术后、慢性肝病、门静脉海绵样改变、胆囊结石。期间口服谷胱甘肽、利胆片等药物治疗。1月前病人肝区疼痛加重,伴头晕、厌油腻,伴腹泻,每天3~4次稀水样便,小便色黄,无腹胀,无恶心、呕吐,就诊于我院感染科门诊。完善肝功能检查:谷丙转氨酶(ALT)45 U/L,谷草转氨酶(AST)66 U/L,谷氨酰转肽酶(GGT)77 U/L,甲胎蛋白(AFP)1.84 μg/L,乙型肝炎病毒DNA<100 kU/L。门诊以肝硬化收入我科。病人自发病以来精神烦躁,饮食可,睡眠欠佳,大小便如上所述,体质量近5年下降约10 kg。家族史:其子腹部超声示肝囊肿,双肾多发囊肿,不除外多囊肾。查体:病人体形消瘦、肝病面容,腹软,右上腹压痛,肝脏触诊质韧,肋下约6 cm,触痛,余查体未见异常。入院后血凝常规检查:抗凝血酶Ⅲ 66%,部分活化凝血酶原时间(APTT) 35.1 s。血常规、尿常规、大便常规、免疫球蛋白、抗核抗体、抗平滑肌抗体、抗线粒体抗体、抗双链DNA、血糖、电解质、肾功能、丙型肝炎抗体、乙型肝炎五项、心电图等检查未见异常。
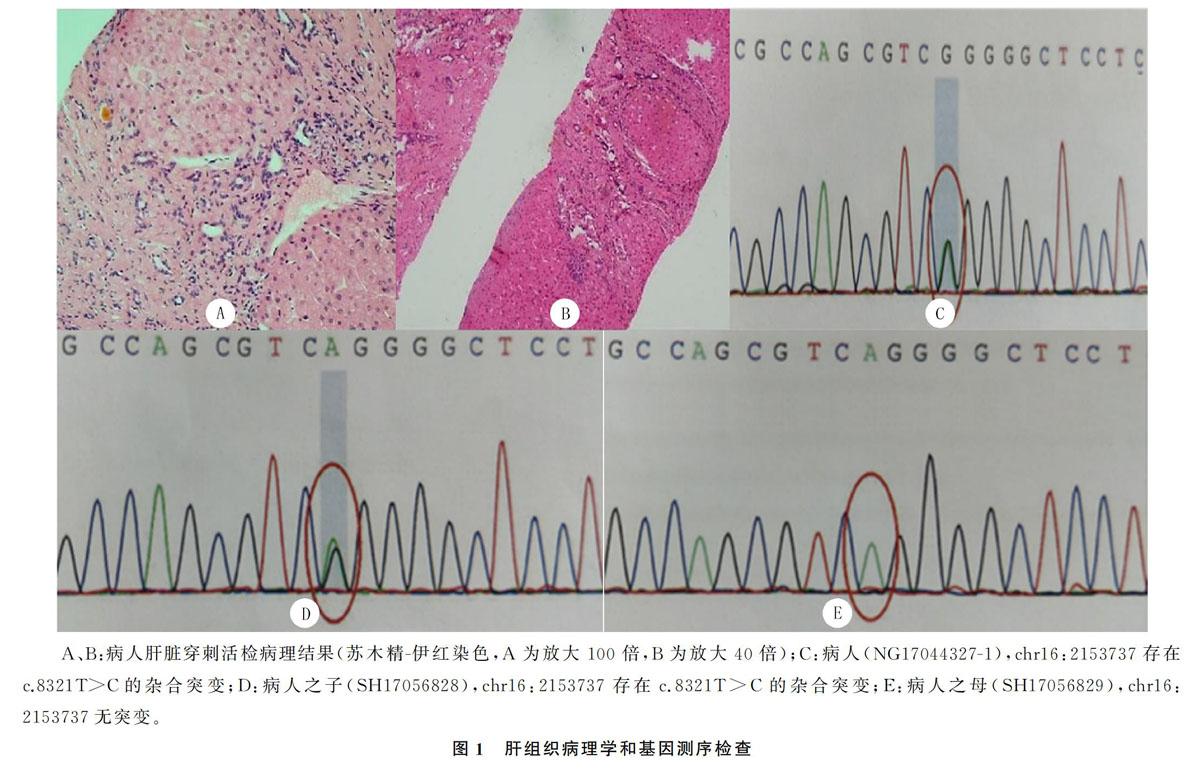
入院后给予保肝治疗,于2017-03-15行超声引导下肝脏穿刺活检术。病理诊断:肝细胞轻度水肿伴淤胆,汇管区肝纤维组织增生明显伴小胆管增生,肝小叶结构破坏,可见纤维组织分隔肝小叶形成的假小叶结构,考虑为结节性肝硬化(图1A、B)。为进一步明确肝硬化原因将病人病理切片外送会诊。①复旦大学附属华山医院病理诊断:肝脏穿刺标本小叶结构凌乱,门管区明显扩大,广泛肝纤维化,宽纤维间隔
内大小不一的小胆管增生活跃,诊断考虑CHF。②金域医学检验中心病理诊断:肝实质由宽窄不一的纤维束分隔成片块状或结节;汇管区少量淋巴细胞、浆细胞、中性粒细胞浸润,局部轻度界面炎,小胆管明显增生,部分形态不规则,管壁周围纤维化,部分小胆管扩张,淤胆;肝细胞轻度水肿,个别肝细胞脂肪变性,可见少量点状坏死,部分肝细胞可见色素颗粒沉着;中央静脉偏位或缺失,支持CHF的诊断。综合上述结果,我院诊断考虑为CHF。其后,病人就诊于中国人民解放军第302医院,将我院肝脏穿刺标本拿到该院病理科会诊,病理结果为:肝组织内假小叶形成,汇管区内大量炎细胞浸润,轻度界面炎症,纤维瘢痕及宽大纤维间隔见大量胆管结构,部分扩张淤胆,诊断为Caroli病伴CHF。并采集病人及其母亲、儿子的外周血送金准医学检验所行基因检测,检测方法为安捷伦外显子芯片捕获+高通量测序,检测产品:SWES_JZ4000plus,结果显示病人样本存在多囊肾,成人1型相关基因PKD1存在一处杂合突变,家系验证结果显示其子存在相同的突变位点,其母无此位点突变;Sanger测序结果显示,病人与其子chr16:2153737存在c.8321T>C杂合突变(图1C~E)。检测产品:MT-04-Blood_线粒体基因组(plus),结果显示该样本线粒体全基因16 569个位点中没有发现已报道明确致病的65种突变。数据解读参考美国医学遗传学和基因组学学院(ACMG)相关指南[2],变异命名参考人类基因组变异协会(HGVS)建议的规则给出。综合病人病史、肝脏穿刺活检及基因检测报告,CHF伴多囊肾诊断明确,Caroli病待诊。考虑病人目前暂无肝功能异常、反复胆管炎、消化道出血等危险,既往已行脾切除相关手术,我院暂未对病人行肝移植等手术治疗,予药物保守治疗。随访至今,病人未出现严重症状,门诊定期复查肝功能、消化系统超声等,较前未有明显变化。
2 讨论
CHF是一种散发性或家族性的常染色体隐性遗传性疾病,于1954年由GRUMBACH首次描述,后于1961年由KERR命名。该病的发病机制至今尚未明确,多数学者认为CHF与DPM相关的肝内胆管发育异常相关[3-4],且有一定的家族遗传倾向,近亲结婚会增加其子女的发病率。CHF发病年龄差异较大,从新生儿到中老年都可发病,但以儿童发病为主[3,5];發病性别差异不明显。根据临床表现的不同,将CHF分为4型:门静脉高压型、胆管炎型、混合型和隐匿型[6-7]。在我国以门静脉高压型为主,主要表现为上消化道出血、脾大、腹水、肝门静脉以及脾静脉扩张、侧支循环开放等,肝功能正常或轻度异常。本文病人肝区不适,查体时发现脾大、门静脉高压,未出现呕血、黑便等上消化道出血表现,且肝功能正常,符合相关文献报道。由于CHF临床表现多样、症状不典型,临床诊断困难。影像学检查如超声、CT、MRI、MRCP等无特异性,但若发现门静脉高压表现、胆管异常或肾脏病变则有助于诊断[8-9]。目前,CHF主要依靠肝组织活检病理确诊,为其诊断金标准。
CHF的病理学表现为正常的肝细胞岛被宽窄不一的成熟的纤维组织分隔,纤维间隔内可见拉长或囊状的成熟的胆
管结构;汇管区与纤维间隔混杂,中心可含正常的或扩张的小叶间胆管;门静脉分支发育不良或缺失,肝细胞岛形状不一,但与中央静脉保持正常的关系;肝板正常,肝实质和纤维间隔内没有明显的炎性改变,无典型的假小叶形成。总结其特征性表现为:汇管区扩大、纤维化,内有小胆管增生,形态各异,但无明显炎症坏死[10-11]。本文病人先后两次行肝脏穿刺活检术,首次肝脏穿刺未明确病因,病理诊断为不明原因肝硬化,其原因可能与穿刺活检组织病理表现不典型、对该病病理认识不足有关,故需提高对CHF的病理表现认识。
本病与Caroli病、多囊肝、胆管错构瘤等在临床及病理上有明显交叉,统称为纤维多囊性肝病。另外,众多学者认为CHF与Caroli病皆与DPM相关,两者事实上是以门静脉周围纤维化及胆管扩张为主要特征的同一疾病的不同阶段,而非单独的或同时发生的疾病[12]。本文病人病理检查可见部分胆管扩张淤胆,于他院病理诊断为Caroli病伴CHF,考虑到其影像学及病理检查未见典型的胆管囊状扩张表现,故Caroli病待诊。有文献报道,超过50%的CHF病人可合并肾脏疾病,以ARPKD最常见,也可合并ADPKD、囊性肾脏发育不良及肾髓质病,但相对少见[1]。本文病人超声及CT等影像学检查可见多囊肾病表现,并且存在PKD1基因突变,为CHF合并ADPKD,相对少见。另外,需要注意的是ADPKD与ARPKD的鉴别,ARPKD是常染色体隐性遗传病,其致病基因为PKD1[13],表现为双肾形成多个进行性增大的囊肿,后期出现肾功能不全[14]。另有研究表明,PKD1亦是CHF及Caroli病的共同致病基因,但本文病人基因检测未见其突变。
在肝移植技术成熟之前,CHF无根治方法,多以针对并发症的治疗为主[15]。对于上消化道出血病人,也可采取内镜下套扎或硬化剂治疗,TIPS或外科分流等[16]。本文病人10年前因脾大、门静脉高压行脾切除术,但并未明确病因。单纯脾切除术虽可以减少1/3的门静脉血流量,降低部分门静脉压力,缓解脾功能亢进,但并不能从根本上解决问题[17]。对于终末期肝病或合并严重并发症病人,肝移植为最佳的根治方法,对于合并肾脏功能损伤病人可行肝肾联合移植[6]。考虑本文病人目前暂无反复胆管炎、肝功能异常、消化道出血等危险,暂未行肝移植等手术治疗,予药物保守治疗。随访至今,病人一般情况良好。
总之,CHF是一种少见的累及肝脏和肾脏的疾病,临床上主要以门静脉高压和肝功能正常为主要特点,肝脏穿刺活检为其诊断金标准。由于该病隐匿性及复杂性特点,极容易误诊、漏诊,当病人出现不明原因门静脉高压且合并多囊肾、肝功能正常或轻度异常时,需要考虑到本疾病。
[参考文献]
[1]JIANG Chang, ZHOU Qiang, JIN Meishan, et al. Congenital hepatic fibrosis with polycystic kidney disease: two case reports[J]. Medicine, 2019,98(20):e15600.
[2]李菲菲,傅兆庆,任万华. 先天性肝纤维化伴Caroli病一例[J]. 中华肝脏病杂志, 2019,27(6):463-465.
[3]AGRWAL S, DABAS A, PAL T, et al. Goldston syndrome with congenital hepatic fibrosis: a rare cause of neonatal cholestasis[J]. Intractable & Rare Diseases Research, 2019,8(2):154-157.
[4]冯茂森,马文斌,汤善宏,等. 先天性肝纤维化的发病机制及诊治现状[J]. 临床肝胆病杂志, 2017,33(3):553-557.
[5]PARKASH A, CHEEMA H A, MALIK H S, et al. Congenital hepatic fibrosis: clinical presentation, laboratory features and management at a tertiary care hospital of Lahore[J]. JPMA. the Journal of the Pakistan Medical Association, 2016,66(8):984-988.
[6]任美欣,韩莹,刘晖,等. 先天性肝纤维化10例临床分析[J]. 北京医学, 2018,40(4):364-365,368.
[7]吴欣,杜霄壤,丁金芳,等. 儿童先天性肝纤维化临床分型及特征[J]. 中国当代儿科杂志, 2016,18(4):335-339.
[8]GUERRA J A, KAMPA K C, ZAPPAROLI M, et al. Congenital hepatic fibrosis and obliterative portal venopathy wi-
thout portal hypertension-a review of literature based on an
asymptomatic case[J]. Arquivos de Gastroenterologia, 2018,55(4):324-328.
[9]田青,袁建军,王绮,等. 超声对Caroli综合征合并先天性肝纤维化、常染色体隐性遗传性多囊肾的诊断价值[J]. 中华超声影像学杂志, 2017,26(6):545-547.
[10]翁成钊,曾晓清,沈小静,等. 先天性肝纤维化三例[J]. 中华肝胆外科杂志, 2018,24(4):274-276.
[11]武健,尹芳,夏琳,等. 13例先天性肝纤维化的临床表现与病理特征分析[J]. 临床肝胆病杂志, 2016,32(3):488-490.
[12]吴欣,吴孟晋,罗生强,等. 先天性肝纤维化和Carolis综合征的临床特征比较[J]. 肝脏, 2015,20(9):667-670.
[13]YANG Ni, LENG Yunji, DAI Shundong, et al. Short article: sequence variations of PKHD1 underlie congenital hepatic fibrosis in a Chinese family[J]. European Journal of Gastroenterology & Hepatology, 2019,31(3):363-367.
[14]曹丽丽,董漪,徐志强,等. 常染色体隐性遗传性多囊肾合并先天性肝纤维化一家系3例报告及文献复习[J]. 临床肝胆病杂志, 2019,35(1):166-168.
[15]YAMADA N, SANADA Y, KATANO T, et al. Pediatric li-
ving donor liver transplantation for congenital hepatic fibrosis using a mothers graft with von Meyenburg complex: a case report[J]. World Journal of Gastroenterology, 2016,22(44):9865-9870.
[16]ZHANG Jinshan, CHENG Wei, LI Long. Laparoscopic distal splenoadrenal shunt for the treatment of portal hypertension in children with congenital hepatic fibrosis: a case report[J]. Medicine, 2017,96(3):e5843.
[17]中華医学会外科学分会胆道外科学组. 胆管扩张症诊断与治疗指南(2017版)[J]. 中华消化外科杂志, 2017,16(8):767-774.
(本文编辑 黄建乡)
[收稿日期]2019-10-29; [修订日期]2020-02-24
[基金项目]山东省医药卫生科技发展计划项目(2018WS379)
[第一作者]王亚平(1993-),女,硕士研究生。
[通信作者]边城(1965-),男,博士,主任医师,硕士生导师。E-mail:jinbeibei@126.com。
