3,5-二硝基-1,2,4-三唑及其复合物感度影响的量化研究*
2020-07-02朱佳平
朱佳平
(广东石油化工学院 化学学院,广东 茂名 525000)
金属的引入可提高含能化合物的爆轰性能,降低其感度,如含铝炸药中引入高密度惰性金属可有效提高其能量[1-3]。作为一种典型的三唑类高能化合物,3,5-二硝基-1,2,4-三唑(DNTz)的感度较高,其离子盐具有良好的稳定性[4]。3,5-二硝基-1,2,4-三唑铵盐的晶体密度ρ为1.632 g·cm-3,热分解温度Tp为170 ℃,特征落高H50为59/80 (#12/12B) cm[5,6]。关于DNTz类含能离子盐的合成报道较多,而理论研究报道较少。本研究结合密度泛函理论B3LYP和微扰理论MP2(full),选用6-311++G(d,p)和6-311++G(2df,2p)基组对DNTz及其复合物(H+、Li+、Na+、K+、Be2+和Mg2+)的电子结构进行全优化,并计算了热引发键C—NO2的解离能BDEC3—N7、Eint(O10…M13)、ρBCP(C3—N7)、E(2)以及qNO2。从微观层面阐述了DNTz及其离子盐感度变化的本质,对其分子设计及合成具有一定的指导意义。
1 计算方法
采用密度泛函理论(DFT)B3LYP/6-311++G(d,p)、B3LYP/6-311++G(2df,2p)和微扰理论MP2(full)/6-311++G(d,p)水平优化了DNTz及其复合物的几何结构,且势能最小(虚频为0)[7,8]。采用MP2(full)/6-311++G**方法计算了复合物的单点能E、键解离能BDE和相互作用能Eint,分析了复合物的自然键轨道NBO,并利用AIM方法研究了其拓扑电荷密度[9]。键解离能BDEC3—N7定义为
BDEC3—N7=BDE(R·)+BDE·NO2…M-BDERNO2…M
(1)
式中:R为3,5-二硝基-1,2,4-三唑自由基,M为H+、Li+、Na+、K+、Be2+和Mg2+,C3—N7表示C—NO2中最弱键。
分子与离子之间的相互作用能(Eint)定义为
Eint=EDNTz…M-EDNTz-EM
(2)
2 结果与讨论
2.1 结构优化
图1显示了DNTz及其复合物的结构和键的临界点,表1列出了复合物的结构参数以及DNTz的晶体数据。由表1可以看出,DNTz分子中C3—N7的键长最大,键级最弱,表明DNTz的热引发键为C3—N7键。三种计算方法的误差在0.0017,0.0015,0.0015 nm,相对误差分别为1.16%,1.03%和1.03%,均在允许误差范围内[10-14]。表明计算方法的选择是可靠的。


图1 DNTz及其复合物的分子结构和键的临界点
由图1可知,除DNTz…Be2+外所有的复合物均为Cs对称性。在B3LYP/6-311++G(2df,2p)计算水平上,复合物DNTz…Li+和DNTz…Na+中O10…M13键长分别为0.2070,0.2416 nm,而杯[4]芳烃锂盐中O…Li+键长为0.1920~0.2089 nm[10,11],杯[4]芳烃钠盐中O…Na+键长为0.2284~0.2337 nm[11,12];表明DNTz与Li+和Na+之间产生相互作用。在MP2(full)/6-311++G**水平上,复合物DNTz…Be2+和DNTz…Mg2+中O10…M13键长分别为0.1617,0.2094 nm,而BeO中O—Be键长为0.1331 nm,MgO中O—Mg键长为0.1749 nm[13]。由此可以看出,O10…M13(M=Be2+,Mg2+)键长均在BeO、MgO的离子半径与其两倍离子半径之间,表明DNTz与Be2+、Mg2+之间发生相互作用。
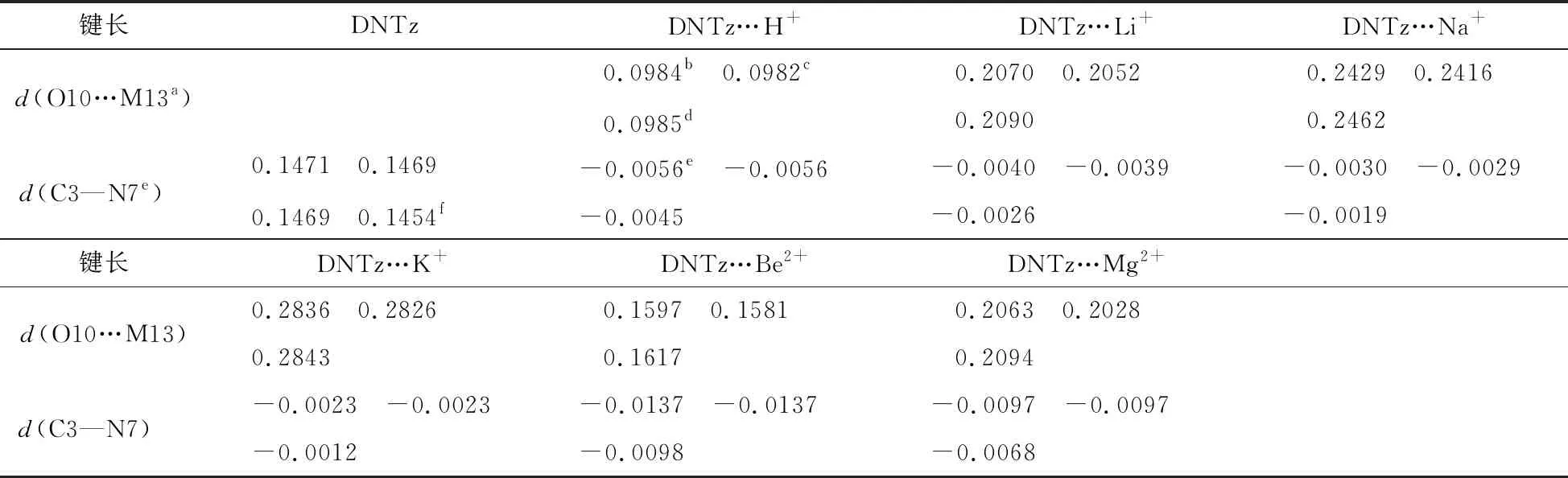
表1 DNTz及其复合物部分键长nm
注:aM13表示相对应复合物中的H+, Li+, Na+, K+, Be2+和 Mg2+;b在B3LYP/6-311++G(d,p)水平;c在B3LYP/6-311++G(2df,2p)水平;d在MP2(full)/6-311++G(d,p)水平;e复合物中C—N键与单体中C—N键的差值;f晶体结构数据。
由表1中的数据可以看出,O10…M13键长顺序为DNTz…Be2+
复合物DNTz…H+中O10—H13在三种计算水平下的键长分别为0.0984,0.0982,0.0985 nm, 接近于H3O+中O—H键长 (0.0980,0.0979,0.0978 nm);表明复合物DNTz…H+中O…H为共价键。
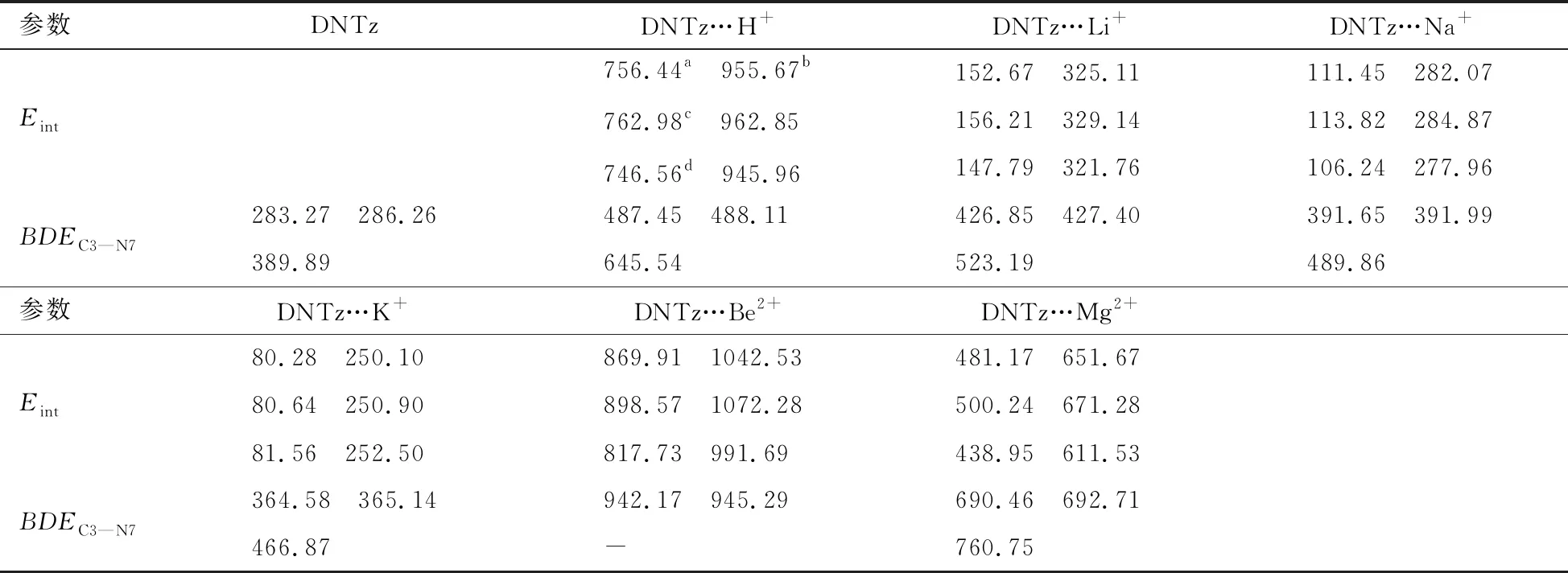
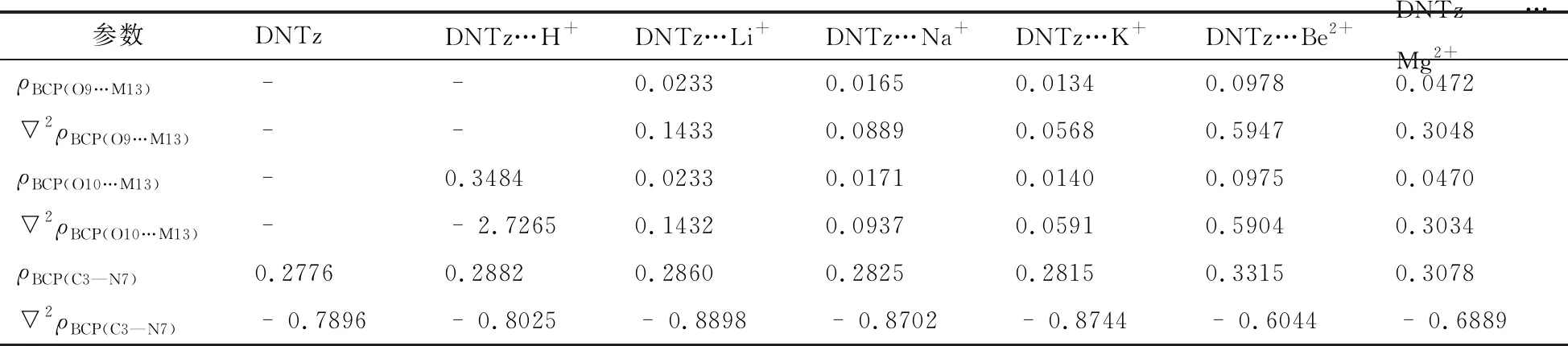
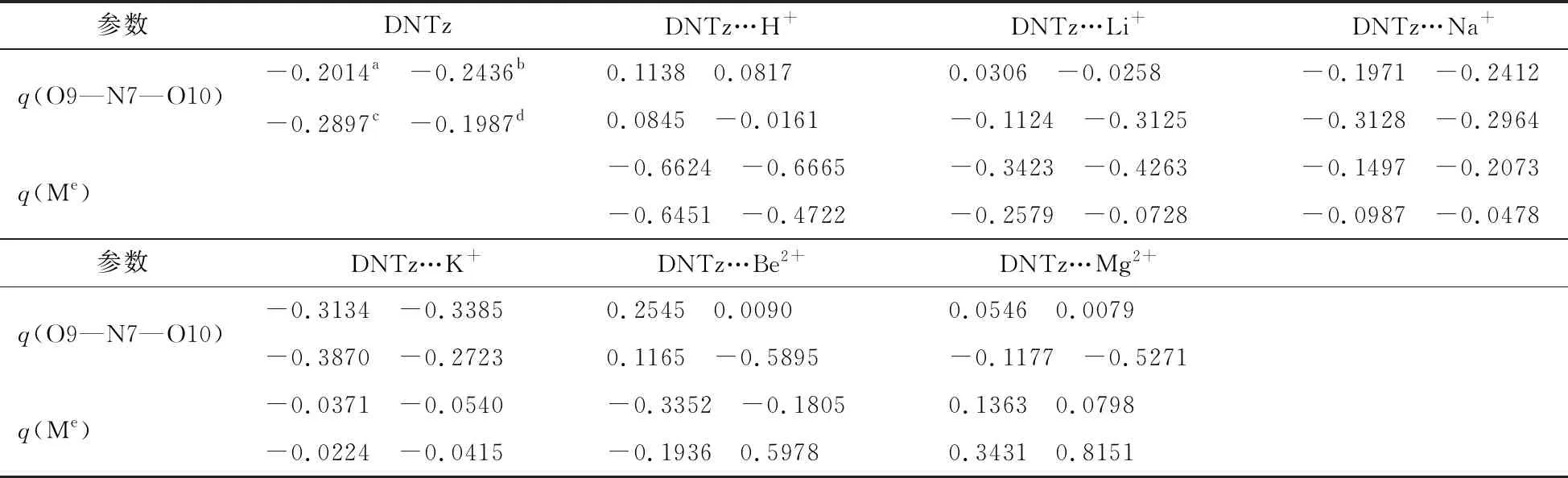
与单体DNTz相比,复合物DNTz…Li+、DNTz…Na+、DNTz…K+、DNTz…Be2+和DNTz…Mg2+中C3—N7键在MP2(full)/6-311++G**水平上分别缩短了 0.0026,0.0019,0.0012,0.0098,0.0068 nm,表明分子与离子之间产生了相互作用,C3—N7键强度增加,使其感度降低[14]。复合物中dC3—N7变量顺序为DNTz…Be2+>DNTz…Mg2+>DNTz…H+>DNTz…Li+>DNTz…Na+>DNTz…K+,而感度顺序为DNTz…Be2+ 表2列出了复合物的Eint以及BDEC3—N7。由表2可看出,在三种计算水平上,DNTz…H+的Eint分别为756.44,762.98,746.56 kJ/mol,接近于3,4-二硝基吡唑体系(DNP…H+, 782.02 kJ/mol[1]);表明复合物DNTz…H+中O10与H+形成了σ键。在B3LYP/6-311++G(d,p)计算水平上,DNTz与Li+、Na+的Eint分别为156.21,113.82 kJ/mol,均接近于杯[4]芳烃锂盐 (205.63 kJ/mol)和钠盐(174.85 kJ/mol)[15];表明DNTz与Li+、Na+产生相互作用。如表2所示,Eint的大小顺序为DNTz…Be2+>DNTz…Mg2+>DNTz…Li+>DNTz…Na+>DNTz…K+,与之前的结构分析结果一致。 表2 DNTz及其复合物中的BDEC3—N7与EintkJ/mol 注:a在B3LYP/6-311++G(d,p)水平下未校正的总能量;b经零点能校正;c在B3LYP/6-311++G(2df,2p)水平下未校正的总能量;d在MP2(full)/6-311++G(d,p)水平下未校正的总能量。 在三种计算水平上,DNTz 中BDEC3—N7分别为283.27,286.26, 389.89 kJ/mol,均接近于文献值[16];表明计算方法的选择是可行的。由表2中的数据还可看出,三种水平下,复合物的BDEC3—N7值均大于DNTz的BDEC3—N7值。特别是DNTz…Be2+,在B3LYP/6-311++G(d,p)和B3LYP/6-311++G(2df,2p)水平上的BDEC3—N7分别为942.17,945.29 kJ/mol,约为DNTz中BDEC3—N7的3倍。即DNTz与Be2+产生相互作用,C3—N7键强度增加,感度随之降低。BDEC3—N7大小顺序为DNTz…Be2+>DNTz…Mg2+>DNTz…H+>DNTz…Li+>DNTz…Na+>DNTz…K+>DNTz,与前面的分析一致。图2为B3LYP/6-311++G(2df,2p)水平下,复合物中BDEC3—N7与dC3—N7成线性关系,如式(3)所示,相关系数为0.9953。 BDE=-4.796×103d+7.305×103 (3) 图3为B3LYP/6-311++G(2df,2p)水平下,复合物中C3—N7键的解离能增量(ΔBDE)与Eint存在线性关系,如式(4)所示,相关系数为0.9986。 ΔBDE=-0.7101Eint+29.184 (4) 表3列出了DNTz及其复合物临界点的ρBCP。从表3中可以看出,除DNTz…Be2+,复合物的ρBCP(O10…M13)值均介于0.0134 ~0.0472 a.u.,接近文献报道值[17];表明DNTz与Li+,Na+,K+,Be2+,Mg2+产生相互作用。由于复合物分子与离子间的拉普拉斯值▽2ρBCP(O9…M13)均为正,表明DNTz分子与离子间产生了闭壳层作用。 图2 C3—N7键解离能与键长的关系 图3 C3—N7键解离能增量与相互作用能的关系 表3 DNTz及其复合物临界点的ρBCP与▽2ρBCPa.u. DNTz…H+中ρBCP(O10…H13)为0.3484 a.u.,其▽2ρBCP值是负值,表明O10…H+为共价键。如表3所示,DNTz…Be2+中C3—N7键的ρBCP最大。ρBCP(C3—N7)大小顺序是DNTz…Be2+>DNTz…Mg2+>DNTz…H+>DNTz…Li+>DNTz…Na+>DNTz…K+,与之前的分析一致。图4为在B3LYP/6-311++G(2df,2p)水平上,复合物的ΔBDEC3—N7与ρBCP(C3—N7)成线性关系(式(5)),其相关系数为0.9981。 ΔBDE=1.1274×104ρBCP(C3—N7)-3.076×103 (5) (6) 图4 C3—N7键解离能增量与电子密度的关系 图5 ΔBDEC3—N7与的关系 表4 复合物C3—N7键与M离子相互作用后轨道能量性质 kJ/mol 表5 DNTz及其复合物中硝基Mulliken电荷以及自然电荷 a.u. 注:a在B3LYP/6-311++G(d,p)水平;b在B3LYP/6-311++(2df,2p)水平;c在MP2(full)/6-311++G(d,p)水平;d在B3LYP/6-311++G(2df,2p)水平;Me表示H+, Li+, Na+, K+, Be2+, Mg2+。 图6ΔBDEC3—N7与分子轨道能的关系 表5列出了DNTz及其复合物中硝基的Mulliken电荷。由表5可知,DNTz中硝基O9—N7—O10的电荷为-0.1987 a.u.。复合物中硝基O9—N7—O10的电荷顺序为DNTz…Be2+>DNTz…Mg2+>DNTz…Li+>DNTz…Na+>DNTz…K+,即其感度顺序为DNTz…Be2+ ΔBDE=1.60848Q+36.65027 (7) 采用密度泛函理论(DFT) B3LYP/6-311++G(d,p)、B3LYP/6-311++G(2df,2p)和微扰理论MP2(full)/6-311++G(d,p)优化了DNTz以及MDNTz (M为H+,Li+,Na+,K+,Be2+和Mg2+)的几何结构,并计算了BDEC3—N7、Eint(O10…M13)、ρBCP(C3—N7),E(2)以及qNO2。结果表明在复合物中C3—N7键长、Eint(O10…M13)、ρBCP(C3—N7)、E(2)以及qNO2均随ΔBDEC3—N7增大而增加,随之感度降低,感度顺序为DNTz…Be2+2.2 能量与稳定性
2.3 AIM分析

2.4 NBO分析




3 结论
