Fast genetic mapping in barley:case studies of cuticle mutants using RNA-sequencing
2020-07-01XiaoFengLiChaoLiQinZhouGuoXiongChenPengShanZhao
XiaoFeng Li ,Chao Li ,Qin Zhou ,GuoXiong Chen ,PengShan Zhao
1.Key Laboratory of Stress Physiology and Ecology in Cold and Arid Regions,Northwest Institute of Eco-Environment and Resources,Chinese Academy of Sciences,Lanzhou,Gansu 730000,China
2.University of Chinese Academy of Sciences,Beijing 100049,China
3.Shanghai Center for Plant Stress Biology,Centre for Excellence in Molecular Plant Sciences,Shanghai Institutes for Biological Sciences,Chinese Academy of Sciences,Shanghai 201602,China
4.Gaolan Station of Agricultural and Ecological Experiment,Northwest Institute of Eco-Environment and Resources,Chinese Academy of Sciences,Lanzhou,Gansu 730000,China
ABSTRACT Barley(Hordeum vulgare L.)is one of the earliest domesticated crop species and ranked as the fourth largest cereal pro‐duction worldwide.Forward genetic studies in barley have greatly advanced plant genetics during the last century;howev‐er,most genes are identified by the conventional mapping method.Array genotyping and exome-capture sequencing have also been successfully used to target the causal mutation in barley populations,but these techniques are not widely adopt‐ed because of associated costs and partly due to the huge genome size of barley.This review summarizes three mapping cases of barley cuticle mutants in our laboratory with the help of RNA-sequencing.The causal mutations have been suc‐cessfully identified for two of them and the target genes are located in the pericentromeric regions.Detailed information on the mapping-by-sequencing,mapping-and-sequencing,and RNA-sequencing assisted linkage mapping are presented and some limitations and challenges on the mapping assisted by RNA sequencing are also discussed.The alternative and elegant methods presented in this review may greatly accelerate forward genetics of barley mapping,especially for labora‐tories without large funding.
Keywords:barley;mapping-by-sequencing;RNA-sequencing;cuticle;point mutations
1 Introduction
The extracellular matrix, namely the cuticle,forms the outermost coat over the aerial surface of al‐most all land plants and serves as a biophysical barri‐er that provides protection against abiotic and biotic stresses(Kosma et al.,2009;Chen et al.,2011a;Lu et al.,2012;Yeats and Rose,2013;Li et al.,2018).The cuticle is also involved in plant development and crop quality improvement(Dominguez et al.,2017;In‐gram and Nawrath,2017).The polymer cutin and nonpolymerized cuticular waxes are two major compo‐nents of the cuticle,while wax compounds are fre‐quently a mixture of very long chain aliphatics and their derivatives, including alkanes, aldehydes, ke‐tones, esters, and primary and secondary alcohols(Jenks et al.,2002;Nawrath,2006;Samuels et al.,2008;Yeats and Rose,2013).A number of genes es‐sential for cuticle biosynthesis and assembly have been characterized in model species Arabidopsis thaliana,for example,Eceriferum 1(CER1)is essential for alkane-forming pathway (Aarts et al., 1995),which have significantly improved our understanding on the molecular mechanism controlling the cuticle biosynthetic pathways(Yeats and Rose,2013;Lee and Suh,2015).Nevertheless,some unique cuticle components are present in many other species(e.g.,β-diketones present in Poaceae species but absent in Arabidopsis)and the genes and enzymes involved in cuticle biosynthesis remain to be identified(Hen-Avivi et al.,2016;Schneider et al.,2016).
Barley(Hordeum vulgare L.)is one of the earliest domesticated crop species and is widely cultivated in diverse environmental regions across the Arctic Circle to the tropics and ranked as the fourth largest cereal production worldwide(Mayer et al.,2012).In addi‐tion to its important agriculture value,barley mutation research has greatly advanced plant genetics through‐out the last century and also has significantly promot‐ed its breeding and improvement(Druka et al.,2011;Mascher et al.,2014).A large number of barley mu‐tants have been created by physical and chemical mu‐tagens since 1928(Stadler,1928).Among these lines,1,580 are categorized as cer mutants with defects on the deposition of epicuticular waxes and have been as‐signed to 79 different cer loci(Lundqvist and Lun‐dqvist,1988).A series of independent recurrent back‐crosses to cultivar(cv.)Bowman have been carried out and 75 cer near isogenic lines(NILs)have been developed(Druka et al.,2011).Further array genotyp‐ing with up to 3,072 single nucleotide polymorphisms(SNPs)defines the chromosomal locations and delim‐its the introgression intervals of these cer NILs(Druka et al.,2011).Recently,some cer mutants were successfully cloned and validated.The cer-c,cer-q,and cer-u mutants exhibit glossy leaf sheaths,inter‐nodes and spikes,and glaucous leaf blades(Schneider et al.,2016).Genetic mapping reveals that these three genes are tightly linked on chromosome 2HS and en‐code a diketone synthase,a lipase/carboxyl transfer‐ase,and a P450 enzyme,respectively,which are cru‐cial for β-diketone biosynthesis(Hen-Avivi S et al.,2016;Schneider LM et al.,2016).The cer-zv and cerym are categorized as spike,leaf sheath,and leaf blade mutants(Figure 1),and cer-yl is a spike and leaf sheath mutant,which have been assigned into three different loci(Lundqvist and Lundqvist,1988).However,bi-parental population mapping results dem‐onstrate that cer-zv,cer-ym,and cer-yl,are allelic mu‐tants with cutin deficiency and the underlying gene is a Gly-Asp-Ser-Leu(GDSL)-motif esterase/acyltrans‐ferase/lipase controlling the deposition of cutin poly‐mer(Li et al.,2013,2015,2017),whereas causal mu‐tations for the dwarf phenotype remain to be resolved.Position cloning of the leaf blade cer-zh mutant re‐veals that a single base change of the β-ketoacyl-CoA Synthase(HvKCS1)results in defects on cuticle load and composition and also changes the germination of barley powdery mildew on leaves(Li et al.,2018).Obviously,a new strategy revolutionizing the tradi‐tional map-based cloning is required to finally unveil the legacy of cer mutants in barley.

Figure 1 Phenotype comparisons of three cuticle mutants with wild type plants.(a)detached leaves of eibi1.b and wild type after 1 h at room temperature,(b)phenotypes of Foma and cer-zv.342 plants,scale bars in both are 10 cm,(c)adult plants of Bowman and cer-b.2,scale bar is 10 cm
The advancement of next-generation sequencing technologies and the well-assembled reference ge‐nome sequences have greatly improved genetic map‐ping strategies(Zou et al.,2016).Many pipelines,such as SHOREmap (Schneeberger et al., 2009),NGM(Austin et al.,2011),and MutMap(Abe et al.,2012),which are based on the combination of classi‐cal bulked-segregant analysis(BSA,Giovannoni et al.,1991;Michelmore et al.,1991)and the whole ge‐nome sequencing,have been developed to identify causal point mutations. Latest released pipelines,such as MutMap+(Fekih et al.,2013)and SIMPLE(Wachsman et al.,2017),work very well with a M2 or M3 segregating population and completely skip the outcross or backcross steps.Some general rules on the bulked population size and coverage per se‐quenced individual are also developed to accurately estimate allele frequency for the BSA(James et al.,2013;Schlötterer et al.,2014).The major obstacle for barley BSA is its huge genome size (5 Gb,Mascher et al.,2017).Alternatively,exome capture sequencing provides a method to reduce the repre‐sentation and complexity of the genome and has been successfully used to mine candidate genes for the many-noded dwarf(mnd)and early maturity 5(eam5)mutants(HvMND/HvCYP78A and HvEAM5/HvPHYTOCHROME C,Mascher M et al.,2014;Pankin A et al.,2014).However,the cost of sequencing load for whole genome sequencing and of exome capture would limit their adoption in barley genetic research,especially in laboratories without large funding.This review summarized three genetic mapping cases in our laboratory using RNA-sequencing(RNA-seq)and the elegant methods could accelerate forward genetics for barley cuticle mutants in the near future.
2 Three mapping cases of barley cuticle mutants using RNA-sequencing
2.1 Eibi1 and mapping-by-sequencing
A spontaneous mutant,namely eibi1,was firstly isolated after two rounds of complete selfing of a wild barley genotype 23-19,which originated from Wadi Qilt in Israel(Chen et al.,2004).This mutant exhibits pleiotropic phenotypes,including dwarf stature,twist‐ed and dark green leaves,more tillers,twisted pedun‐cle,less spike size,and reduced fertility(Figure 1a).Most significantly,eibi1 mutants are hypersensitive to water stress and its relative water loss rate of detached leaves is higher than most of other wilty mutants,such as tomato flacca mutant(Chen et al.,2004).Three crosses against the cultivar(cv.)Morex,23-19(a near-isogenic line[NIL]of the eibi1),and another wild genotype 23-73 from Wadi Qilt were constructed and segregation analyses confirmed that eibi1 pheno‐type is controlled by a single recessive mutation(Fig‐ure 2a;Chen et al.,2004,2009a).Thirty-four informa‐tive SSR markers were used to target the chromosome location of the eibi1 and genotyping results of 43 F2mutants from a cv.Morex×eibi1 population(43F2)show that the eibi1 is closely linked with Hvm60 and Bmag603 markers on chromosome 3H(Figure 2b;Chen et al.,2009a).Further genotyping of two newly established populations,cv.Morex×eibi1(155F2)and 23-73×eibi1(108F2),with SSR markers delimit‐ed the eibi1 into a 7.2 cM interval between Bmag828 and Bmag603,which is located in a pericentromeric region(Figure 2c;Chen et al.,2009a).All known CAPS markers and ESTs located on chromosome 3H were then explored to genotype these two popula‐tions. The candidate intervals were not narrowed down,however,a EST AV922032 was found to be cosegregated with the eibi1 in the 23-73×eibi1(108F2)population although this marker was not informative in the cv.Morex×eibi1(155F2)population.EST AV922032 has a rice homologue(Os01g0188100)on chromosome 1 and barley ESTs based on syntenic in‐formation were thus developed as potential markers.Three ESTs, including CB859971, BF263820, and AV918546,were completely linked with the eibi1 in the 23-73×eibi1(108F2)population,while the inter‐val was delimited into 1 cM between Bmag828 and AV918546 in the cv.Morex×eibi1(155F2)popula‐tion.Clearly,EST AV918546 is co-segregated with the eibi1 in both populations(Figure 2c;Chen et al.,2009a).Barley-rice syntenic information near EST AV918546 was exploited again to identify potential markers.The genetic interval was finally delimited from 0.11 cM to 0.10 cM with genotyping individuals increased up to 1,682 in barley and the physical inter‐val was also narrowed from 112.8 kb to 76.5 kb in rice genome(Chen et al.,2009b,2011b).Among the rice collinear segment,ten candidate genes were in‐cluded and one gene encoded an ABCG31 transporter which was chosen as the candidate for the eibi1(Fig‐ure 2c;Chen et al.,2011b).Genetic analyses based on another four eibi1 allelic mutants and two rice osabcg31 mutants validated the eibi1 phenotype and expression experiments confirmed that the Eibi1 mRNA is highly accumulated in the elongation zone(EZ)at the three-leaf stage of barley(Chen et al.,2011b).Detailed analyses of the cuticle phenotype re‐vealed that the underlying gene of the eibi1,Hv-ABCG31,functions as an ABCG full transporter for the correct cutin matrix formation and in turn contrib‐ute to water conservation in barley leaves(Chen et al.,2011a,2011b).Similar function of OsABCG31 is also found in rice and these results suggest that the ABCG31 protein is essential for land plant coloniza‐tion and hence for terrestrial life evolution(Chen et al.,2011a;Garroum et al.,2016).

Figure 2 Conventional mapping and mapping-by-sequencing of eibi1.(a)segregation analyses,(b)chromosome location,(c)rough and fine mapping results,(d)flowchart of mapping-by-sequencing of eibi1.The data in(a),(b),and(c)are from previous publications(Chen et al.,2004,2009a,2009b,2011),while the data in d are from Zhou et al.(2017a).Markers in red color denote the cosegregation with the eibi1 locus
The genetic mapping process of the eibi1 is timeconsuming and laborious(Chen et al.,2004,2011b).Recently,RNA-seq has been implemented to re-map‐ping the eibi1 mutation(Zhou et al.,2017a).The EZ tissue of the third leaf was sampled at three-leaf stage from the NIL 23-19 and homozygous eibi1 seedlings.Two independent libraries were developed and se‐quenced using Illumina HiSeq 2000 platform.Subse‐quently,reads from the 23-19 and eibi1 libraries were separately assembled and then clustered together to generate a non-redundant reference dataset(78,799 unigenes;Figure 2d,Zhou et al.,2017a).Initial SNP calling using SOAPsnp program yielded 13,785 SNPs from 4,070 unigenes.Because of characteristics of the 23-19 and eibi1 seedlings,only homozygous variants were considered and four SNPs(C/T,G/A,G/A,and T/A)were finally identified from four different unige‐nes (Unigene14072_All, Unigene19902_All, Unigene46051_All, and Unigene47049_All; Zhou et al.,2017a). Gene annotation revealed that Unigene19902_All encodes an ABC transporter G family member protein and the mutation G/A changes a tryp‐tophan to a stop codon(W->*),which is consistent with the amino acid change identified by the conven‐tional forward mapping(Chen et al.,2011b).Three other genes encoding 1-acyl-sn-glycerol-3-phosphate acyltransferase 4,RNA ligase isoform 1,and ribosom‐al protein,are not related to cuticle development.These results suggest that mapping-by-RNA-seq is an alternative and elegant method for gene identification in barley,concerning its large genome size.
A major concern when using mapping-by-RNAseq for screening causal SNP is that the sequencing depth is largely different among samples and the tran‐script of the expected gene may not be detected due to low or no expression(Hill et al.,2013;Miller et al.,2013).The drought hypersensitive phenotype of the eibi1 was easily observed at a very early growth stage,even for the first expanded leaf(Chen et al.,2009b). Moreover, cutin deposition mainly occurs during the period of cell elongation when the EZ is de‐veloping (Richardson et al., 2007; Chen et al.,2011b).In Zhou et al.(2017a),the EZ tissue of the third leaf from seedlings at three-leaf stage were used and the sampling time is early enough to cover the ex‐pression window of the candidate gene.Indeed,there were 801 unique reads mapping against the Unigene19902_All in the eibi1 library,although the expres‐sion was reduced more than two fold in the eibi1 mu‐tant(Zhou et al.,2017a).Another application of RNAseq is to develop potential markers.Six out of eight EST markers,which were used to delimit the eibi1 in‐terval into 1 cM(Chen et al.,2009a),have been vali‐dated by comparison of eibi unigenes and cv.Morex contigs while the remaining two markers are located in the introns(Zhou et al.,2017a).
2.2 Cer-zv and mapping-and-sequencing
The cer-zv.268 was firstly isolated from the ethyl‐ene imine-induced cv.Foma and then a NIL cer-zv.268(BC4F3, GSHO2207) was constructed by repeated backcrossing to the cv.Bowman(Figure 1b).A clear Mendelian 3:1 ratio for the leaf tests was exhibited in F2progenies from both 23-19×cer-zv.268 and cerzv.268×OUH602 populations(Figure 3a;Li et al.,2013).Bi-parental 23-19×cer-zv.268(30F2)popula‐tion mapping showed that cer-zv.268 is closely linked with a length polymorphism marker AK364271 and lo‐cated near the centromeric region of chromosome 4H(Figure 3b;Li et al.,2013).A set of 23 informative markers derived from a barley high-density transcript linkage map(Sato et al.,2009)and a high-throughput SNP map(Close et al.,2009)were subsequently em‐ployed to delimit the cer-zv.268 locus into 10.8 cM and 1.7 cM intervals by using 23-19×cer-zv.268(93F2)and cer-zv.268×OUH602(88F2)populations,respec‐tively,and AK251484 was found to be co-segregated with the cer-zv.268 in both populations(Figure 3c;Li et al.,2013).Since four EST markers(AK359440,AK252593,AK251484,and AK363409)showed cosegregation with the cer-zv.268 in 23-19×cer-zv.268 population,only cer-zv.268×OUH602 population was used for next fine mapping(Li et al.,2017).An ex‐panded population(3,682F3)with 17 EST markers fur‐ther narrowed the candidate interval into 0.028 cM in the centromeric region(Li et al.,2017).Collinear com‐parison between EST mapping markers and the barley genome zipper information(Mayer et al.,2011)re‐vealed that the gene order is not very consistent and the possibility that a great number of genes might be re‐combinationally locked into this small genetic interval cannot be ruled out,and identification of the cer-zv.268 is thus not feasible using the conventional bi-parental mapping(Li et al.,2017).
The cer-zv,cer-ym,and cer-yl mutants were cate‐gorized as three independent loci(Lundqvist and Lun‐dqvist,1988).However,the phenotypes of deficient cuticle in these mutants,such as water loss rate of de‐tached leaves and permeability to toluidine blue stain‐ing,are very similar(Li et al.,2013,2015,2017).Ar‐ray-based genotyping of NIL cer-ym.753 and ceryl.187 have delimited the expected loci into two large intervals but with an overlap on the chromosome 4H(Druka et al.,2011).Bi-parental mapping against the wild barley OUH602 showed that both cer-ym.753(123F2)and cer-yl.187(96F2)cosegregate with the marker AK364461 on the chromosome 4H and pair cross tests further confirmed the allelism of cer-zv.268,cer-ym.753,and cer-yl.187(Li et al.,2015,2017).The target interval(1 cM)of cer-yl.187 also harbors the candidate intervals of cer-zv.268(0.028 cM)and cer-ym.753 (0.045 cM) (Figures 3d-3e; Li et al.,2017).These results suggest that a same locus is shared among cer-zv.268,cer-ym.753,and cer-yl.187 mutants,at least for the deficient cuticle phenotype.
Detailed analyses of cutin and wax content in cerzv.268 and cer-ym.753 mature leaves have revealed that the accumulation of six major cutin monomers is reduced to a similar level in these two mutants(Li et al.,2013,2015,2017).Partially expanded third leaves of Foma(T1)and cer-zv.342(T2)seedlings were thus sampled for RNA-seq,since the cutin deposition in barley leaves mainly starts at this growth stage(Rich‐ardson et al.,2007;Chen et al.,2011b).Unigenes were assembled independently and variants including SNPs and insertions/deletions(Indels)were screened using the blastn program(Figure 3f;Li et al.,2017).A total of 363 variants in 263 unigene pairs were iden‐tified.A comparison of these unigenes versus the ES‐Ts located into a larger region(42.45−48.72 cM)on the Genome Zipper chromosome 4H(Mayer et al.,2011),which totally covers the cer-zv.268 interval(AK370363-AK251484;Figure 3c),was performed and eight candidate SNPs were identified in five unigene pairs.Further screening based on the gene an‐notation identified two candidate genes for the cer-zv mutants.Sanger resequencing of these two genes in two cer-zv lines,two cer-ym lines,and four cer-yl lines finally confirmed that mutations in a GDSL-motif esterase/acyltransferase/lipase gene are responsi‐ble for the cuticle deficiency in cer-zv,cer-ym,and cer-yl leaves(Figure 3f;Li et al.,2017).
2.3 Cer-b.2 and RNA-seq assisted linkage mapping
The monofactorial recessive mutant cer-b.2(GSHO434; GSHO1081), exhibiting a glossy spike and leaf sheath phenotype,originated from the X-ray induced cv.Maja(PI 184884)and the causal locus is roughly targeted on chromosome 3HL (Figure 1c;Franckowiak et al., 2016). An NIL line BW107(BC6F3,GSHO1989)was generated by a series of backcrosses of cer-b.2 into cv.Bowman and array ge‐notyping have delimited the introgression interval of cer-b.2 into 24.09 cM between marker 2_1513 and 1_0754(Druka et al.,2011).BW107,namely BW-NIL in Zhou et al.(2017b),was then crossed against the wild genotype 23-19 to construct the bi-parental map‐ping population and a Mendelian 3:1 ratio of the glau‐cous to glossy leaf sheath was observed again in F2progenies(Figure 4a;Zhou et al.,2017b).Phenotype examination revealed that epicuticular wax crystal structure of BW-NIL is different with that of 23-19 and the total amount of deposited wax is also less on the surface of the BW-NIL plant.Wax composition analy‐ses showed that 14,16-hentriacontanedione(β-diketone)and some unknown compound syntheses are blocked while primary alcohols and aldehydes are highly accu‐mulated in BW-NIL plants(Zhou et al.,2017b).
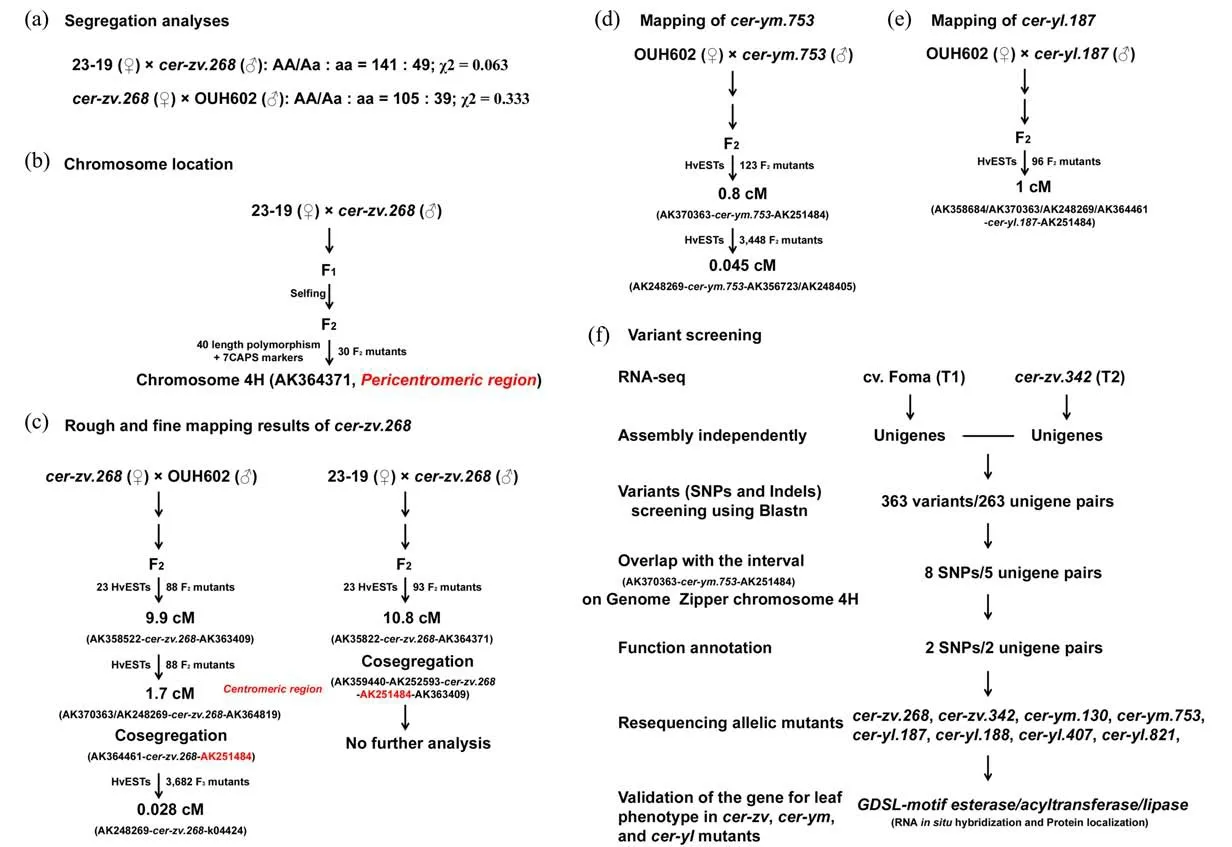
Figure 3 Mapping-and-sequencing of cer-zv,cer-ym,and cer-yl.(a)segregation analyses,(b)chromosome location,(c)rough and fine mapping results of cer-zv.268,(d)mapping results of cer-ym.753,(e)mapping results of cer-yl.187,(f)flowchart of mapping-and-sequencing and validation of cer-zv,cer-ym,and cer-yl.The data in a-e are from previous publications(Li et al.,2013,2015,2017),while data in(f)are from Li et al.(2017).Markers in red color denote the cosegregation with the cer-zv.268 locus
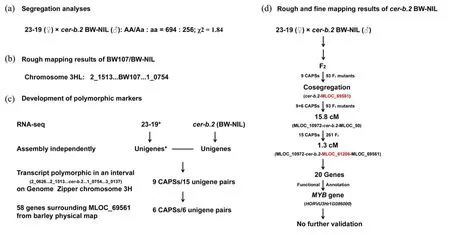
Figure 4 RNA-seq assisted linkage mapping of cer-b.2 BW-NIL.(a)segregation analyses,(b)rough mapping results of cer-b.2 BW107/BW-NIL,(c)polymorphic markers are developed by the comparison of 23-19 and cer-b.2 BW-NIL transcripts located in the candidate interval on the genome zipper chromosome 3H(Mayer et al.,2011)and the region surrounding MLOC_69561 from the barley physical map(Mayer et al.,2012),(d)rough and fine mapping results of cer-b.2 BW-NIL,CAPSs markers used for mapping are from the description in(c).The data in(a),(c),and(d),are from Zhou et al.(2017b),while data in(b)are from Druka et al.(2011).Markers in red color denote the cosegregation with the cer-b.2 BW-NIL locus.To be noted,the unigene dataset of 23-19 is from Zhou et al.(2017a)
To identify the sequence polymorphisms within the parent lines,total RNA from the partially expand‐ed third leaf of BW-NIL was used to develop the tran‐script dataset by RNA-seq(Zhou et al.,2017b).Bar‐ley genes located in a larger region between marker 2_0626 and 3_0137,which includes the introgression interval of cer-b.2 on the Genome Zipper chromo‐some 3HL(Druka et al.,2011;Mayer et al.,2011),were employed to isolate the corresponding tran‐scripts from the de novo assembled transcriptome da‐tasets of BW-NIL and 23-19(Zhou et al.,2017a,2017b).A set of nine markers derived from the poly‐morphic transcripts between two parental lines were then used to genotype the 23-19×BW-NIL(93F2)population and MLOC_69561 was found to be com‐pletely linked with the cer-b.2(Zhou et al.,2017b).The gene information based on the barley physical map(Mayer et al.,2012)was further exploited to de‐velop polymorphic markers between two parental lines in the same way.A total of 15 markers surround‐ing MLOC_69561,including six newly developed,were used to genotype the 23-19×BW-NIL(354F2)population and the cer-b.2 locus was finally targeted into a 1.3 cM interval flanked by MLOC_69561 and MLOC_10972(Figure 4b;Zhou et al.,2017b).This interval includes 20 genes according to the latest re‐leased barley genome(Mascher et al.,2017)and one MYB family gene(HORVU3Hr1G086000)is suggest‐ed as the candidate of the cer-b.2(Zhou et al.,2017b).
3 Summary and perspectives
Forward genetic screens in barley have identified a number of genes essential for agronomic traits,such as Nud for the covered/naked caryopsis phenotype(Taketa et al.,2008),Eibi1 for the cuticle develop‐ment(Chen et al.,2011b),and Non-brittle rachis 1(Btr1)and Btr2 for the grain dispersal(Pourkheiran‐dish et al.,2015).However,conventional mapping of barley mutants is usually labor intensive,partially due to its relatively complex genome.The development of array genotyping(Druka et al.,2011)and exome-cap‐ture sequencing(Mascher et al.,2014;Pankin et al.,2014) has significantly accelerated the process of mapping mutations in barley genome by analyzing multiple genetic markers simultaneously, but these techniques have not been widely adopted because of the high costs associated with array preparation and exome capture.Transcriptome is smaller in size and less complex than the whole genome.RNA-seq can generate greater read depth with fewer reads in com‐parison with the whole-genome-sequencing.This re‐view presents three mapping cases assisted by RNAseq in barley and the causal mutations have been suc‐cessfully identified for two of them,namely Eibi1 and Cer-zv.268,which are located in the pericentromeric region.Similarly,RNA-seq is also used to identify the causal mutation underlying the six-row spike3(vrs3)phenotype by sequencing two allelic mutants simulta‐neously(Van Esse et al.,2017).
Accumulating evidence has demonstrated that cu‐tin deposition in the epidermis of the barley leaf oc‐curs at a very early growth stage,even before the time when the EZ of the third leaf is formed,whereas wax deposition is delayed and occurs gradually with the leaf development(Richardson et al.,2007;Chen et al.,2011b).To increase the likelihood of sequencing the causative gene,partially expanded third leaves,a special development window when the cuticle pheno‐types commence,are sampled in all three cases to cap‐ture the mutant transcript.This is also a key to in‐crease the odds of successful mapping with the help of RNA-seq.
Thanks to the power of RNA-seq,we can now identify the point mutations using single sets of se‐quencing libraries.However,there are still some limi‐tations and challenges of mapping with the help of RNA-seq. Identification of the differentially ex‐pressed genes can be limited using a single RNA-seq replicate (Anders and Huber, 2010). For example,Zhou et al.(2017a)found 269 genes are differentially expressed in eibi1 mutants,whereas no homologs of the known cuticle-related genes are included.Given the cutin deficiency phenotype of the eibi1 mutant,the number of differentially expressed genes might be underestimated.To accurately estimate the effect of eibi1 mutation on the expression levels of genes,three separate biological replicates should be included in fu‐ture studies.Dwarfism is another important pheno‐type in cutin mutants eibi1,cer-zv,cer-ym,and cer-yl(Chen et al.,2004;Li et al.,2013,2015;Franckowiak et al., 2016). Allelic mutants of eibi1 and rice osabcg31 mutants all exhibit strong dwarfism and de‐tailed analyses have revealed that a dysfunctional cuti‐cle may affect the signaling network of plant growth and defense (Chen et al.,2011b;Garroum et al.,2016).In cer-zv.268,dwarfism is co-segregated with the cutin phenotype(Li et al.,2013).Both cer-ym.753 and cer-yl.187 plants are semidwarf but not as severe as that of cer-zv.268(Li et al.,2015;Franckowiak et al.,2016).One possibility is that the dwarf phenotype is an indirect effect of mutations in GDSL-motif esterase/acyltransferase/lipase and variations in plant height are caused by different mutations in the GDSLlipase.Similarly,the rice mutant wilted death lethal(wdl)has a T-DNA insertion mutation in a GDSL-lipase gene resulting in a dwarf phenotype and plant death at seedling stage(Park et al.,2010).Mutants of cer-zv.268,cer-ym.753 and cer-yl.187 retain donor parent markers in the same region of 4HL(Druka et al.,2011),however,allelic tests have shown two con‐trast results:mutations at these loci are not allele(Franckowiak et al.,2016)and are allele(Li et al.,2017).Since our previous mapping was mainly fo‐cused on the leaf water loss at seedling stage,the dwarf phenotype may not be co-segregated with cuti‐cle phenotype in some mapping populations and the possibility that another mutation is hiding in the cen‐tromeric region of chromosome 4H thus cannot be ruled out.In case of mapping the dwarfism,RNA could be extracted from a number of different tissues or development stages.Increasing the breadth of tran‐scripts captured and the sequencing depth would be very helpful to identify the underlying gene for the dwarf phenotype.Alternatively,different allelic mu‐tants of cer-zv,cer-ym,and cer-yl could be analyzed simultaneously.Increasing the number of allelic mu‐tants is expected to improve sensitivity for the final candidate genes and address the discrepancy in allelic tests and the mutations responsible for the dwarfism.
4 Conclusion
Barley has a large genome.This paper presents three methods, including mapping-by-sequencing,mapping-and-sequencing, and RNA-sequencing as‐sisted linkage mapping,which can be used for target‐ing the position of causal genes on barley chromo‐somes.These methods are cost-saving and efficient and can also be extended to mutation mapping in oth‐er species with a large genome.
Acknowledgments:
This research was funded by grants from the National Natural Science Foundation of China(No.41621001,No.31870381,and No.31970352)and by the Youth Innovation Promotion Association,CAS(2018463).
杂志排行

Sciences in Cold and Arid Regions的其它文章
- A meta-analysis of the impacts of forest logging on soil CO2 efflux
- Quantitatively estimate the components of natural runoff and identify the impacting factors in a snow-fed river basin of China
- Mapping the dynamic degree of aeolian desertification in the Shiyang River Basin from 1975 to 2010
- Numerical simulations on cutting of frozen soil using HJC Model
- Editors-in-Chief Yuanming Lai and Ximing Cai
- A modified numerical model for moisture-salt transport in unsaturated sandy soil under evaporation