染色体6p25 缺失综合征 1 例报告并文献复习
2020-06-30厉广栩
潘 翔 逯 军 厉广栩 陈 振
中南大学湘雅医学院附属海口医院儿科(海南海口 570208)
染色体6 p 25 缺失综合征临床罕见,是由包含FOXC 1、RIPK 1、SERPINB 6、TUBB 2 A、TUBB 2 B等基因在内的6 号染色体短臂发生部分缺失引起的临床综合征。2005年经荧光原位杂交(fluorescence in situ hybridization,FISH)技术及比较基因组杂交(comparativegenomic hybridization,CGH)技术验证2例临床表现为智力低下、眼部异常、听力丧失及特殊面容患儿的染色体6p25区带存在缺失[1]。本文分析1例染色体6p25缺失综合征患儿的临床资料,并复习相关文献。
1 临床资料
患儿,男,2岁9个月,因抽搐入住海口医院儿科。患儿于体温38.0 ℃时出现抽搐,表现为呼之不应,双眼上翻,面色苍白,口唇发绀,牙关紧闭,四肢强直抖动,持续约5分钟后自行缓解,抽后精神疲倦,无呕吐、腹泻、咳嗽及喘息等。入院体格检查:体温37.8℃,呼吸26次/min,脉搏122次/min,体质量12 kg,身高85 cm;神清,精神疲倦,浅表淋巴结无异常;特殊面容,前额突出、眼距宽、低鼻梁;牙齿缺损,牙釉质发育不良,流涎;心、肺、腹无异常;四肢肌张力可,腹壁反射、提睾反射、膝跳反射均可引出,脑膜刺激征阴性,巴宾斯基征阴性。患儿系G2P2,足月顺产,出生体质量3.4 kg,出生时无窒息史。运动发育与同龄儿相似,语言发育落后,现仅能喊“爸爸、妈妈”。既往有反复呼吸道感染史。父母非近亲结婚,父亲体健,智力正常;母亲智力低下、精神障碍,有多次抽搐史,亦有听力下降和蛛网膜囊肿病史。实验室检查:血尿粪常规、肝肾功能、电解质、体液免疫及细胞免疫指标大致无异常;外周血染色体核型分析无异常。头颅磁共振成像(MRI)示右侧颞极蛛网膜囊肿,胼胝体发育不良及第三、四脑室扩张(图1)。眼科显微镜下检查示双侧角膜直径近12 mm(较正常同龄儿宽);双角膜周边见角膜后胚胎环,近全周,以鼻侧和颞侧更为明显(图2);双侧虹膜色素异常;右侧瞳孔呈水滴状向颞下移位,符合Axenfeld-Rieger综合征(Axenfeld-Rieger syndrome,ARS)改变。听力测试无异常。脑干听觉诱发电位(brain stem auditory evoked potential,BAEP)检测示10~15 Hz时分别多次给予50~100 db刺激,双侧波形分化不良(图3)。彩色多普勒超声心动图检查无异常。

图1 患儿头颅MRI 表现

图2 眼科显微镜下发现角膜后胚胎环形成

图3 脑干听觉诱发电位异常表现
为明确发病原因,经医学伦理委员会审核以及监护人知情同意,采集患儿外周静脉血2 mL,送深圳安吉康尔医学检验实验室对患儿进行全外显子测序检测,生物信息学分析过程中利用国际千人基因组计划(1000 Genome Project)、基因组聚合数据库(Genome Aggregation Database)、外显子组整合数据库(Exome Aggregation Consortium)等注释变异在人群当中的频率,利用在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM),人类基因突变数据库(Human Gene Mutation Database,HGMD),与疾病相关的人类基因组变异数据库(ClinVar)等进行疾病注释,利用PolyPhen2、SIFT、MutationTaster等软件进行蛋白功能损伤预测分析。根据美国遗传学与基因组学学会指南(ACMG)进行筛选分析,未发现疑似致病点突变。拷贝数变异(copy number variation,CNV)分析:利用专利WEAVER算法,采用滚动基线、亲缘(Kinship)分析、负二项分布(NB)估算每个外显子的基线分布模型,估测样本每个染色体层面的整倍性(Ploidy),再估测样本每个外显子层面拷贝数从的概率分布,采用马尔可夫随机场(Markov Random Field),动态更新每个外显子的最终拷贝数,分析发现患儿6号染色体p25.3~p25.2(chr 6:393140-3226909)存在一段2.833 M的杂合缺失(图4)。
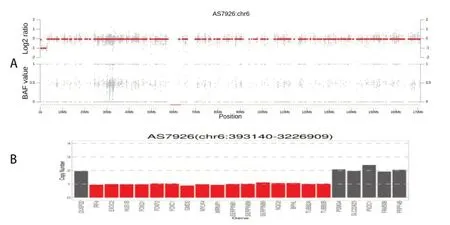
图4 患儿基因检测结果
患儿入院后予抗感染、镇静止痉、维持水电解质平衡等对症支持治疗后未再发生抽搐,生命体征平稳。由于患儿父母拒绝行基因检测,故不能确定患儿染色体所缺失片段的来源。
2 讨论
利用关键词“6p25 deletion syndrome”和“6p25缺失综合征”分别检索英文数据库(PuBMed、Scopus和Elsevier)和中文数据库(万方医学网、中国知网和维普中文科技期刊),累计检索到68篇相关英文文献,未检索到关于本病报道的中文文献。通过系统分析文献后发现,染色体6p25缺失综合征罕见但临床表现多样,其发生与遗传获得或自发突变有关,缺失片段越大,受累基因越多,患者临床表现越复杂。该病的病例报道涉及缺失片段大小从0.9 kb至6.7 Mb不等,大部分病例报道存在FOXC1、SERPINB6、TUBB2A、TUBB 2 B基因受累,临床表现以眼前节发育障碍、青光眼、耳聋、语言发育迟缓、骨骼发育异常、特殊面容及神经系统发育异常较为常见[1-11]。本例患儿6号染色体p25.2~p25.3区域发生2.833 Mb大小的片段缺失,在缺失片段的16个基因中,经查阅并分析文献后发现,与患儿临床表现密切相关的基因有FOXC1、SERPINB6、TUBB2A和TUBB2B。
FOXC1为叉头框(forkhead box,FOX)转录因子基因家族成员之一,该基因家族编码的转录因子参与个体的发育过程,其中包括胚胎发育、组织细胞分化和某些病理过程(如肿瘤)[12]。FOXC1编码的转录因子对中胚层、神经嵴和眼部发育至关重要[13],其缺失在染色体6p25缺失综合征临床表现中扮演着重要角色。目前与FOXC 1缺失有关的文献报道以ARS 最为多见。ARS 是一种罕见的常染色体显性遗传病,发病率为1/200000。儿童ARS病例报道较成人更少见,以眼前节发育障碍和其他系统异常为特征,其中眼前节受累包括前房角结构异常、角膜和虹膜异常等,而其他系统受累则包括颅面畸形、牙齿发育不全、听力下降和心脏发育异常等。本例患儿双侧角膜直径较正常同龄儿宽、双侧虹膜色素异常、右侧瞳孔移位以及双侧角膜后胚胎环形成均符合ARS 中的眼前节发育障碍表现;患儿亦存在ARS的其他系统异常,如前额突出、眼间距过宽、低鼻梁、牙齿发育不全及脑干听觉诱发电位波形分化异常,但彩色多普勒超声心动图检查未发现心脏发育异常的证据。ARS 有三种类型:Ⅰ型与PITX 2基因异常有关,Ⅱ型的致病基因不详,Ⅲ型与FOXC1基因异常有关[14],其中与FOXC1异常有关的ARS 演变为青光眼的可能性较大[15]。有文献报道约半数的ARS 患者最终发展为青光眼[16],故需长期随访以监测眼内压变化。与染色体6p25缺失综合征有关的耳聋基因除FOXC1外,还有SERPINB6,两者均可导致感音性或传导性耳聋,且具有渐进性和年龄依赖性的临床特点。虽然目前患儿听力测试无异常,但脑干听觉诱发电位存在异常,故仍需长期随访以监测患儿听力变化[17-18]。
微管蛋白为球形蛋白质,按结构不同可分为三种:α-微管蛋白、β-微管蛋白和γ-微管蛋白,均在神经元增殖、神经元迁移和迁移后生长环节发挥关键作用。本例患儿6 号染色体所缺失的两个基因TUBB2A和TUBB2B与β-微管蛋白有关。有文献报道,TUBB2A和TUBB2B异常在神经系统可表现为脑回发育不良、小脑发育畸形、脑室扩张、胼胝体发育不良及癫痫发作等,临床表现差异性较大[19-20]。根据头颅MRI 检查结果,提示本例患儿存在第三、四脑室扩张和胼胝体发育不良,与国外文献报道部分吻合。
患儿6号染色体p25.3~p25.2区域发生2.833 Mb的杂合缺失,结合患儿母亲智力低下、抽搐、听力下降和蛛网膜囊肿的病史,不排除患儿的基因缺失来源于其母亲的可能,但患儿父母拒绝行基因检测,故无法进一步验证。
综上,受缺失片段大小和缺失基因种类等因素影响,染色体6p25缺失综合征的临床表现差别很大。产前咨询和产前诊断对保证优生优育至关重要。以抽搐、发育落后并伴有特殊面容就诊的患儿,应警惕染色体异常的可能。外周血染色体核型分析是可作为初步筛查的一种手段,但对于染色体微缺失的检出,仍需进一步完善基因检测以及时干预,改善预后。
