DNA 4mC 甲基化修饰位点预测的研究进展
2020-06-30许召春刘华军
许召春 刘华军
(景德镇陶瓷大学信息工程学院,江西 景德镇333403)
最常见的DNA 甲基化修饰分别是N6- 甲基腺嘌呤(6mA)、5- 甲基腺嘌呤(5mC)、N4- 甲基胞嘧啶(4mC)。DNA 6mA 和5mC 位点广泛存在于原核生物和真核生物中,而DNA 4mC 位点只存在于原核生物中[1]。4mC 于1983 年被发现,是细菌DNA中最不常见的甲基化DNA 碱基[2]。DNA 4mC 在限制性修饰体系中起着重要作用[3]。为了更好地理解它们的功能机制,识别4mc修饰是非常重要的。DNA 4mC 位点的实验筛选是耗时、费力和昂贵的。因此,开发生物信息学工具大规模准确高效地识别4mC 位点是湿实验的有效补充。近年来研究者们基于机器学习方法研发了一系列高效的4mC 位点高通量识别方法,本文就4mC 位点预测研究进行综述,并对此提出展望。
1 现有4mC 预测器
1.1 iDNA4mC
iDNA4mC[4]是最早由陈伟与林昊两个团队利用机器学习方法共同提出的预测4mC 修饰位点的预测工具。含4mC 位点的阳性样本是从MethSMRT 数据库中获取,涉及线虫、果蝇、拟南芥、大肠杆菌、嗜碱菌和地杆菌六个物种,最终采用滑窗法(最优窗口长度为41bp)构建了高质量的平衡数据集(表1)。

表1 基准数据集物种正样本及负样本数量分布
DNA 样本序列由核苷酸物理化学属性和核苷酸密度进行编码,每个核苷酸被转化为4 维离散型向量,采用支持向量机作为分类器,在六种物种数据上执行jackknife 交叉验证,主要的性能评价指标ACC 与MCC 值见表2。
1.2 4mCPred
4mCPred[5]预测器是由邹权团队开发,沿用了iDNA4mC 预测器的训练样本数据,为了充分提取基准数据集中的信息,利用三核苷酸的位置特异性偏好和电子- 离子相互作用的伪电位值将DNA 序列转化为数值向量。为了获得更好的泛化预测模型,采用最优特征选择技术(F-score)来选择最优特征子集。作者尝试了不同的分类算法,包括朴素贝叶斯、KNN、随机森林及SVM,最终基于SVM构建了具有更好性能的分类模型,jackknife交叉验证结果见表2,结果表明,相比于iDNA4mC 预测器,4mCPred 预测器的整体性能有一定程度提高。
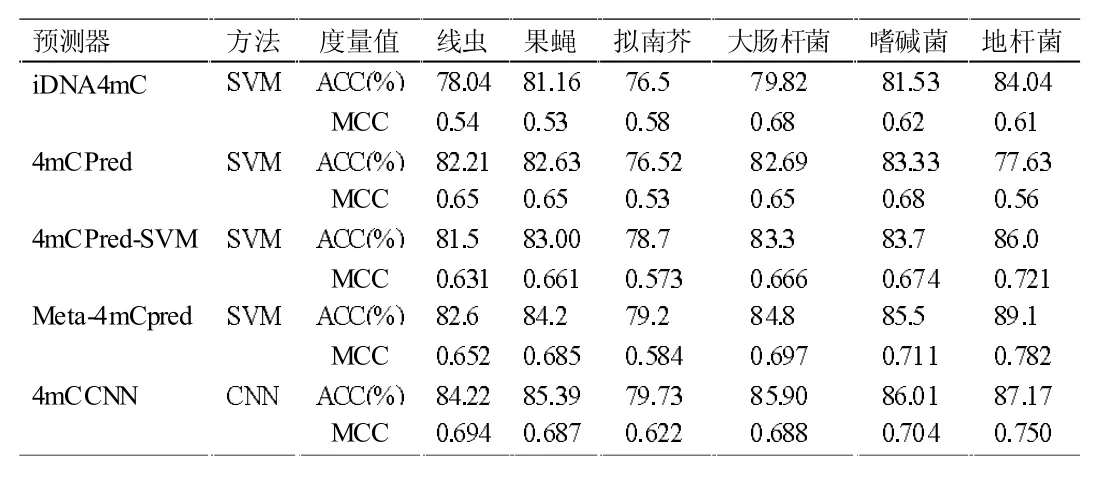
表2 六种物种数据集上各类预测器交叉验证结果
1.3 4mcPred-SVM
在前两个预测器的基础上,邹权团队充分利用基于序列信息的特征表示算法提出了新的预测器4mcPred-SVM[6],用于DNA 4mC 位点的全基因组检测。为了提高特征表示能力,作者采用了两步特征优化策略,从而获得最具代表性的特征。利用所得到的特征和支持向量机(SVM)自适应地训练不同物种的最优模型,结果详见表2。对6 个物种的基准数据集的比较结果表明,与最先进的预测器相比,预测器4mcPred-SVM 能够在预测4mC 位点方面获得更好的性能。重要的是,基于序列的特征能够可靠而稳健地预测4mC 位点,有助于发现潜在的重要序列特征,用于预测4mC 位点。
1.4 Meta-4mCpred
Manavalan 等人[7]采用了一种特征表示学习方案,基于4 种不同的机器学习算法和7 种不同的特征编码,生成了56 个概率特征,涵盖了不同的序列信息,包括成分信息、物理化学信息和位置特定信息。然后,利用概率特征作为支持向量机的输入,最终建立Meta-4mCpred 预测器。交叉验证结果表明来自上述6 个不同的物种的Meta-4mCpred 的总体平均准确率为84.2%,这比现存最好的预测器高出大约2%-4%(见表2)。
1.5 4mCCNN
KHANAL 等人[8]基于上述六种物种的相同基准数据集,采用较为流行的one-hot 编码,利用卷积神经网络开发了4mCCNN预测模型。性能最好的超参数是通过使用网格搜索方法获得,交叉验证结果显示,4mCCNN 预测器性能相比前几个预测器更加良好(见表2),这也意味着深度学习算法在特征表征方面更具优势。
1.6 4mCpred-EL
虽然基于机器学习方法在其他物种中有很好的4mC 鉴定前景,但是目前还没有一种方法可以用于检测小鼠基因组中的4mC 位点。Manavalan[9]提出了一种新计算方法,称为4mCpred-EL,是识别小鼠基因组中4mC 位点的第一个方法,其中使用了四种不同的机器学习算法和七个特征编码方法。然后将这些特征编码的预测概率值作为特征向量,再一次输入到机器学习算法中,将相应的模型通过集成学习进行融合决策,结果见表3。
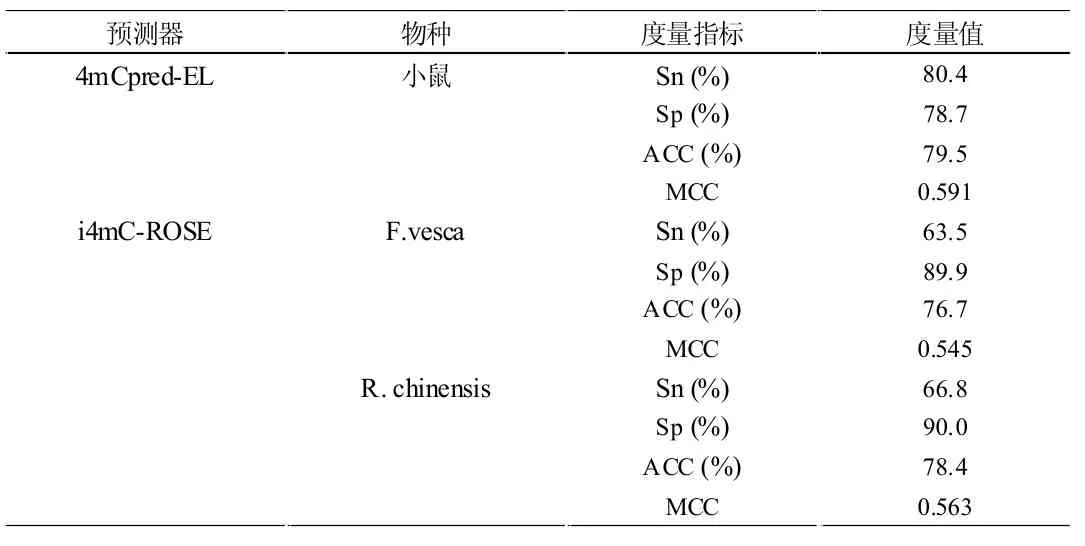
表3 其他物种基准数据集上预测器性能
1.7 iEC4mC-SVM
虽然上述基于机器学习的DNA 4mC 位点的预测器总体性能较好,能提供研究者对4mC 修饰的生物学功能和机制更深入的了解,但是,现有的识别大肠杆菌4mC 位点的分类器性能仍然有待提高。为此,一种新的基于SVM的4mC 位点预测模型iEC4mC-SVM被LV 等人提出,该模型采用多特征融合,并结合光梯度增强机特征选择技术(LGBM)选择最优特征子集,结果比最新的大肠杆菌性能更高,具体度量值见表3。
1.8 i4mC-ROSE
MehediHasan 提出了一种新的预测因子i4mC-ROSE,用于确定蔷薇科中F. vesca 和R. chinensis 基因组中的4mC 位点。首先,利用随机森林(RF)算法分别联合k- 空间光谱核苷酸组成(KSNC)、电子- 离子相互作用伪电位(EIIP)、k-mer 组成(Kmer)、二进制编码(BE)、二核苷酸理化性质(DPCP)和三核苷酸理化性质(TPCP)特征表示方法,生成六个概率分值。其次,将六种概率得分与线性回归模型相结合,提高预测性能。文献表明,i4mC-ROSE 是第一个预测蔷薇科基因组中4mC 位点的计算工具。
为了方便广大研究学者进行DNA 4mC 修饰位点预测分析,除了iEC4mC-SVM 预测器没有在线预测功能,基本每个研究团队都开发了用户友好的在线预测器,用户可通过表4 所提供的链接直接免费访问在线预测器。

表4 在线预测器访问链接
2 展望
最近几年,DNA 4mC 修饰位点预测方面已做了大量的研究工作,并取得了相当不错的成绩,但是仍然存在一些局限性,主要体现在以下几个方面。首先,用于模型构建的训练样本没有更新,大多数预测器仍然是基于首套数据而构建。其次,所采用的分类算法大体还是以传统分类算法SVM为主,只有4mCCNN采用了深度学习中的卷积神经网络CNN。再者,从预测结果上看,预测结果还有一定的提升空间。未来这方面的工作可围绕着这些问题开展,扩大数据集规模,增加物种数量,创建新的特征表示方法,利用深度学习算法进一步提高4mC 位点预测精度,以更好地理解4mC 位点的功能机制。
