高温质子交换膜燃料电池性能衰减机理与缓解策略
——第一部分:关键材料
2020-06-29王子乾杨林林孙海
王子乾,杨林林,孙海
(1 中国科学院大连化学物理研究所醇类燃料电池及复合电能源研究中心,辽宁大连116023;2 大连洁净能源国家实验室,辽宁大连116023;3 中国科学院大学,北京100049)
当今社会正处于经济高速发展的时代,然而经济的发展也带来了严重的能源与环境问题,2017年全球一次能源消耗量达到了惊人的13511.2 百万吨油当量,其中化石能源占比高达85.2%[1],相对应的,2017 年全球CO2排放量也达到了创纪录的3.7×1010吨[2],其中电力产业与交通运输业的CO2排放占比高达65%[3]。为了缓解当前严峻的环境压力,急需优化现有的能源利用模式,大力推广低碳燃料并推进工业、运输电气化的进程。
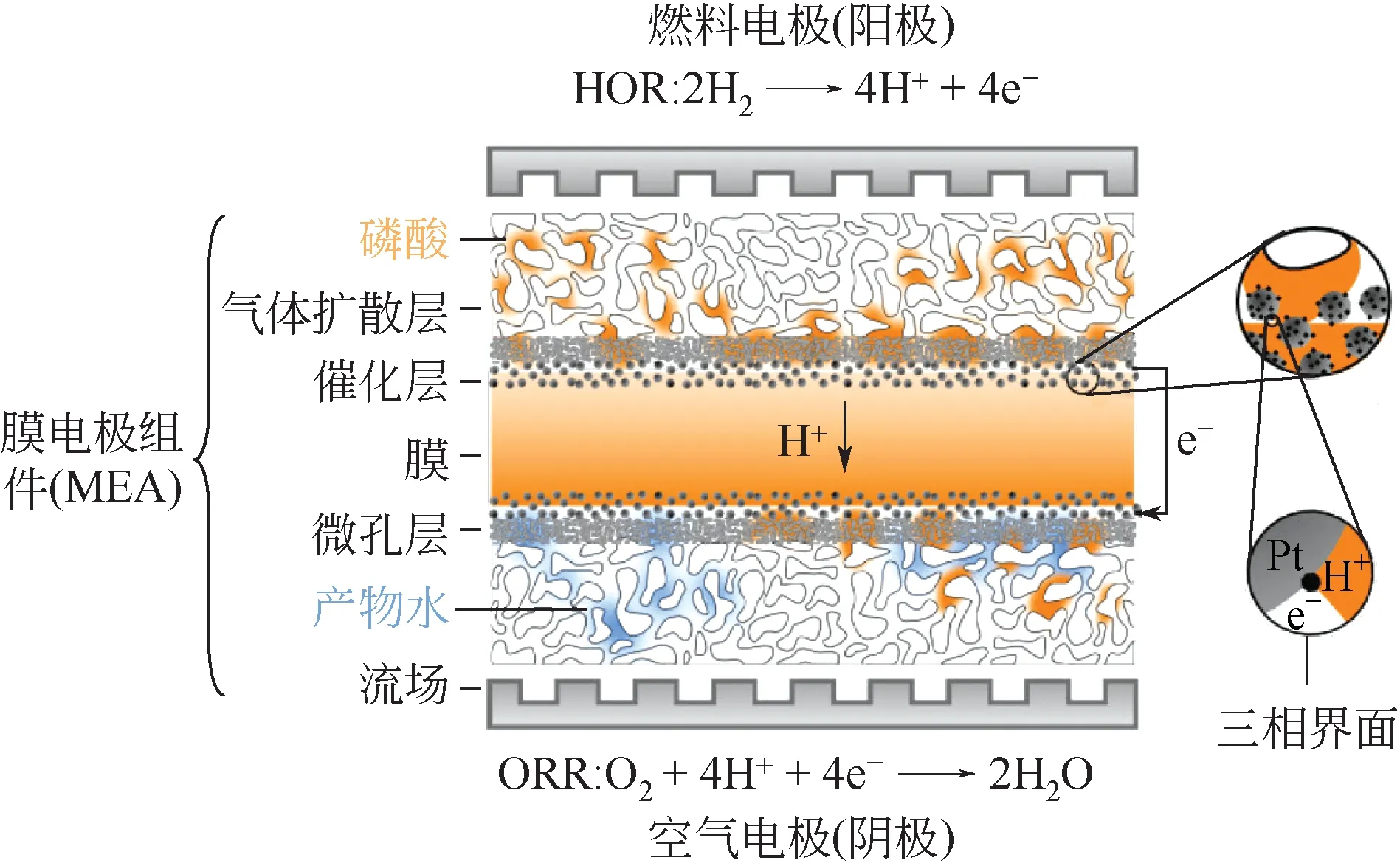
图1 HT-PEMFC工作原理以及结构示意图[5]
质子交换膜燃料电池(proton exchange membrane fuel cell,PEMFC)是近几十年发展起来的一种新型能量转换装置,其发展时间较长、技术较为成熟。由于现有Nafion 膜本身的特性,PEMFC 的工作温度通常在80℃附近,这使得其耐CO 毒化的能力极差,并且PEMFC 的水热管理较为复杂,这些缺点都给PEMFC 的应用推广带来了一定的困难。为了解决这些问题,近些年研究人员开发了一种基于磷酸/聚苯并咪唑(polybenzimidazole,PBI)的高温质子交换膜燃料电池(HT-PEMFC),其工作原理以及结构如图1所示。HT-PEMFC工作温度通常为150~180℃,这大大增加了其耐CO 毒化的能力[4],同时该温度下无液态水生成,且尾气废热利用价值更高,因此HT-PEMFC 具有燃料选择范围广、系统简单、综合能量利用率高等优势,其在车载电源、便携式电源、固定电站以及微型热电联产系统等领域均表现出了巨大的潜力。
然而HT-PEMFC 各组件长时间处于高温酸性环境中,其面临的膜降解、催化剂团聚、碳载体腐蚀以及双极板腐蚀等问题比PEMFC 严重。目前,实验室中报道的HT-PEMFC 稳态运行测试时间最高为17800h[6],动态运行测试最高为4042h(1562次启停循环)[7],这与美国能源部(DOE)制定的目标还有一定的差距,如表1所示,因此提升电池组件的耐久性仍是该领域目前的研究重点。
1 高温质子交换膜性能衰减机理与缓解策略
1.1 高温质子交换膜材料特性
目前HT-PEMFC常用的PBI膜是一类含有苯并咪唑基的芳香杂环聚合物,其具有优异的热稳定性、化学稳定性和机械强度,在HT-PEMFC 中主要起隔绝两侧电极以及传递质子的作用。其中质子在膜内的传递主要依赖于膜内游离磷酸构成的氢键网络,如图2 所示,该传递机制被称为Grotthus 质子传递机制[9]。一般而言,膜内磷酸含量的增加有利于构建质子传递路径,从而提高膜内质子电导率。但是需要注意的是,磷酸的引入也会削弱聚合物分子链之间的相互作用,从而引起膜的膨胀,并导致机械强度的大幅降低,因此在膜的制备过程中通常会根据膜材料与制备工艺选用适中的磷酸含量。

表1 2020年DOE燃料电池寿命目标[8]

图2 Grotthus质子传递机制示意图[9]
自1995 年Wainright 等[10]首次在HT-PEMFC 领域报道了PA/PBI 膜后,研究人员相继开发了大量基于PBI 以及其衍生物的聚合物膜,如图3 所示。PBI聚合物膜的性质和合成方法在前人的综述中都有较为详尽的描述[11-12],这里不再过多阐述。
1.2 高温质子交换膜性能衰减机理
聚合物膜的降解与电解质的损失是高温质子交换膜性能衰减或失效的主要原因之一,如表2所示。
1.2.1 机械损伤

图3 常见的PBI以及其衍生物分子结构
膜发生机械损伤的原因主要与受力有关。在膜电极热压以及电池组装过程中,错误的操作或过高的装配压力可能会导致膜结构受损。在电堆运行过程中,PBI膜在两侧双极板长时间的压力作用下会发生蠕变,从而导致膜变薄或产生微裂纹,其中流道界面或密封边缘等机械应力不均匀的区域更容易产生穿孔或剪切撕裂[13]。此外在电堆运行过程中,膜内磷酸浓度与分布会随电流密度、温度以及湿度的变化而发生改变,进而引起膜的膨胀或收缩,从而导致应力发生变化[14]。Maier等[15]报道,当电流密度从0 增加到0.140A/cm2时,膜电极组件(MEA)发生了约20%的体积膨胀。同样,膜电极反应区与非反应区之间的膨胀差异也会产生局部应力变化[16],这种局部的应力变化可能会加速膜内裂纹和微孔的产生。除了上述提到的挤压形变,阴阳两极过高的气压差也可能会造成膜穿孔,从而导致膜失效。

表2 HT-PEMFC膜降解与磷酸流失方式
1.2.2 热降解
热降解指高分子材料由热作用引起的降解,一般而言,PBI膜具有良好的热稳定性,只有当温度高于600℃时,PBI 分子链上咪唑环与亚苯基连接的碳与胺氮(NH)之间的化学键才容易断裂,从而发生聚合物骨架的断裂与膜结构的崩塌[17],因此正常运行过程中HT-PEMFC 不会发生明显的热降解。然而需要注意的是,当温度大于140℃时,膜内磷酸倾向于发生脱水缩合,形成多聚磷酸,如式(1)所示。
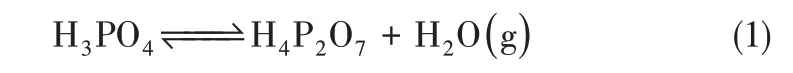
这种自脱水现象往往会导致膜内质子电导率的下降[18]以及膜机械强度的变化。在实际运行过程中,电化学反应产水能够与磷酸自脱水相抵消,但是当局部温度过高时,膜内质子电导率的下降会直接影响电池的性能,同时局部膜机械强度的变化还会增加膜受到机械损伤的可能性。
1.2.3 化学降解
化学降解指高分子材料受化学因素(例如氧化自由基攻击、酸碱水解等)的影响而发生的降解,其中氧化自由基(例如HO·和HOO·)的攻击被认为是PBI膜发生化学降解的主要原因[19]。
(1)自由基生成机理 通常氢气或氧气的渗透会导致氧的不完全还原,从而产生H2O2[20],如式(2)~(4)所示[9]。

这些生成的H2O2可能会进一步反应生成氧化自由基,然而目前关于膜内H2O2反应生成氧化自由基的机理并没有定论。多数人认为这些自由基的形成来源于H2O2与过渡金属离子(例如Fe2+、Cu2+)的反应[21],如式(5)和式(6)所示。

这一机理在采用芬顿试剂的加速应力测试(AST)中得到了很好的验证,然而在实际操作过程中,过渡金属离子的来源是一个值得注意的问题,目前认为其可能来自于端板、管道等金属部件的腐蚀或含过渡金属元素杂质的溶解。
此外,Zhao等[22]则认为铂催化剂本身就可以催化H2O2分解产生氢氧自由基,甚至活性炭也能加剧这一过程,其机理如图4所示,该机理不涉及过渡金属离子,但目前难以定量分析其对PBI膜化学降解的影响程度。
(2)膜化学降解机理 PBI 膜的化学降解过程较为复杂,早先Chang 等[23]通过傅里叶变换红外光谱与核磁共振分析后认为,自由基的氧化攻击导致了与咪唑环相连苯环的分裂,随后形成了二羧酸和奎宁基团,如图5所示。

图4 铂催化过氧化氢生成自由基的反应机理[22]
Liao 等[24]利用尺寸排阻色谱、扫描电镜和傅里叶变换红外光谱等方法,提出了端点断裂与中点断裂两个机理来解释聚合物膜的氧化降解过程,如图6所示。其中端点断裂机理指在膜表面,端点处的羧基受自由基的攻击发生氧化,随后氧化过程沿着分子链进行直到形成稳定的末端氨基,这一过程导致了聚合物的质量损失。而中点断裂机理指在聚合物膜内部,自由基攻击苯并咪唑与亚苯基连接的碳原子,经过一系列反应使得咪唑环开环,并最终导致聚合物主链断裂,生成新的端胺基与羧基,这一过程导致了聚合物分子量的降低。此时新生成的末端羧基会加剧前者的氧化过程,从而加剧聚合物的质量损失。
然而值得注意的是,上述这些机理均建立在芬顿实验的基础上,在实际操作过程中,有研究显示PBI 膜的氧化降解过程会受到磷酸的抑制[25-26]。其中Liao 等[25]提出了3 种可能原因:一是磷酸与过渡金属离子的络合,抑制了过渡金属离子的反应活性;二是磷酸降低了pH,增加了过氧化氢稳定性;三是磷酸与聚合物氨基的相互作用阻碍了自由基的攻击。此外Chang 等[26]还认为PO·的形成会抑制·OH的活性。
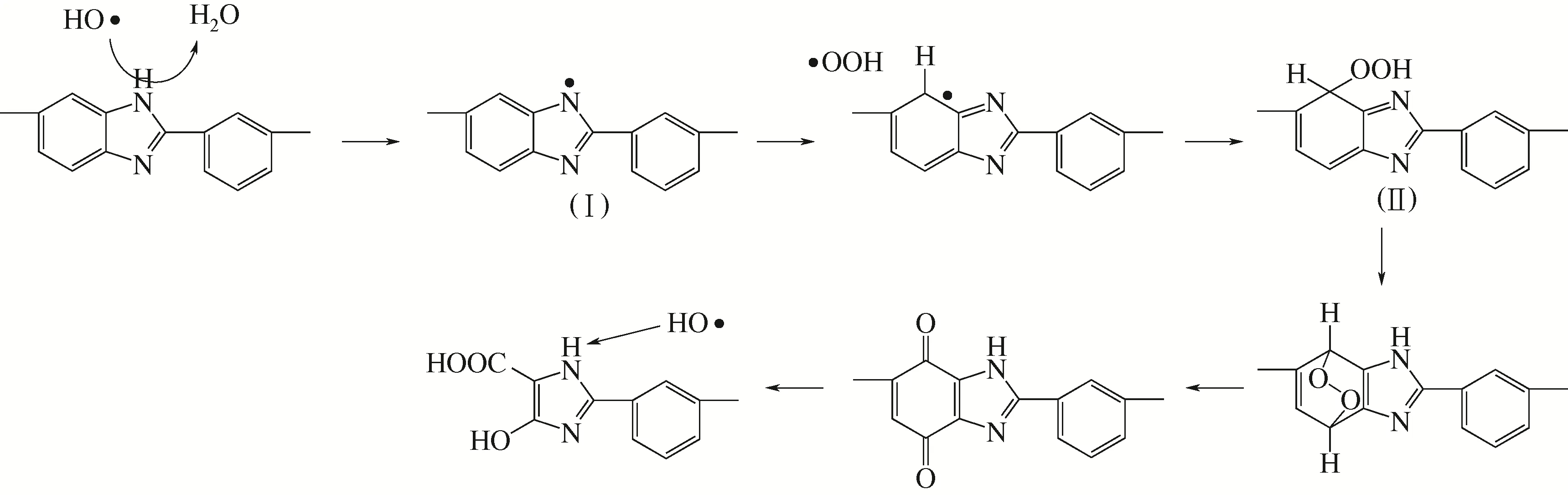
图5 Chang等[23]提出的PBI膜化学降解机理

图6 Liao等[24]提出的PBI膜化学降解机理
近期Ossiander等[27]提出了一种关于膜降解引起性能衰减的新观点,该观点认为PBI分子链在磷酸中存在溶解迁移会导致电池性能的降低。由于磷酸浓度在催化层中的分布因水的产生而呈现梯度,而PBI分子在磷酸中的溶解性伴随磷酸的稀释和聚合物链长的增加而降低[6],因此当PBI 小分子链迁移至磷酸浓度较低的微孔层时,可能会发生沉淀并堵塞孔隙,导致传质阻力增加;而稍大的PBI分子链可能会在催化剂表面析出,从而降低其电化学活性面积(ECSA)。
1.2.4 磷酸损失
通常PBI膜内初始磷酸含量很高,在电池寿命初期,大量的磷酸会在膜电极热压以及电堆装配过程中溢出流失[28],在随后的活化过程中,MEA内的磷酸含量会逐步趋于稳定。在电池寿命后期,磷酸的过量损失往往会导致膜内质子电导率的降低以及三相界面的减少,从而引起电池性能的衰减。
(1)磷酸随液态水流出 早先Li等[29]发现,由于磷酸具有较强的亲水性,PBI膜内非键合的磷酸在液态水或甲醇溶液的冲洗下会迅速流失,这意味着当HT-PEMFC 的运行温度低于100℃时,电池内部产生的液态水可能加速磷酸的损失,从而引起电池性能的迅速衰减[11]。此外,磷酸的流失还会降低PBI 膜的断裂伸长率,从而导致PBI 膜更容易发生断裂。因此,为了避免液态水的生成,HT-PEMFC的运行温度通常在120℃以上。
(2)磷酸蒸发 磷酸蒸发是HT-PEMFC 正常运行过程中磷酸损失的主要途径,目前提出的蒸发机理主要有两种:一是酸气溶胶机理[30],即磷酸形成雾状或液滴状的气溶胶随气流排出,该机理最早提出于磷酸燃料电池(PAFC)领域,并被用来解释PAFC 的高磷酸损失率;二是蒸汽蒸馏机理[28],即通过水蒸汽蒸馏的方式促进磷酸蒸发,该机理被认为是HT-PEMFC磷酸蒸发的主要机理。
实验中评价磷酸蒸发损失量的常用方法是分析阴阳极尾气中的痕量蒸发酸。近些年的实验研究显示,磷酸损失速率与温度、湿度、气体流速以及电流密度等因素密切相关。其中磷酸蒸汽压与温度呈指数关系[31],如图7所示,因此温度的提升会显著增加磷酸的损失速率;湿度的增加能促进聚磷酸重新生成正磷酸,从而增加磷酸的蒸汽压[32];电流密度的影响与湿度相似,如图8所示,然而由于磷酸迁移的存在,电流密度的改变还会影响到磷酸在MEA中的分布,从而加速了磷酸损失[33];气体流速的影响主要与出口处磷酸蒸汽压有关,如图9 所示,若出口处的磷酸蒸汽压达到饱和,则此时的磷酸损失速率与气体流速成正比,若未达到饱和,则总磷酸损失速率随气体流速增加而增加,但单位体积废气中磷酸含量降低[31]。Søndergaard 等[33]进一步将气体总体积与电池性能衰减相关联发现,当通入的气体总体积达到某一阈值时,电池性能开始迅速衰减,这一现象支持了磷酸损失会限制电池寿命的观点。然而由于在收集磷酸前存在冷凝、双极板吸酸以及电池组件腐蚀等过程,通过尾气收集法测得的磷酸损失量偏低,这可能导致研究者错误判断磷酸损失带来的影响。Jakobsen等整理了前人的一些实验数据,发现不同研究组测得的磷酸损失速率可以相差数个数量级,这显然是一个需要重视的问题[34]。鉴于尾气收集法的种种缺陷,Søndergaard等[33]建议使用酸碱滴定测量MEA 中剩余的磷酸来评估整体的磷酸损失。此外,还建议在研究操作参数的影响时,需要考虑该操作条件对水含量以及局部温度的影响,这些因素均会干扰最后的结论。

图7 磷酸蒸汽压与温度关系图[31]
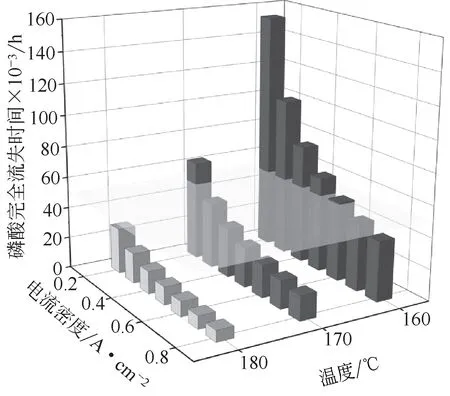
图8 磷酸损失与电流密度/温度关系图[31]

图9 磷酸损失与电流密度/气体流率关系图[33]
(3)双极板吸酸 传统石墨板的高孔隙率通常会导致部分磷酸被双极板所吸收,这些磷酸可能会加剧双极板孔隙的扩张和亲水性的增加[35]。Matar等[36]报道,过量的磷酸不仅会导致磷酸滞留在双极板内,并且还会导致双极板表面出现以C、P、O元素为主的固体团聚物。Pilinski等[37]的测量结果显示,在160℃、0.3A/cm²的条件下,恒流放电下430h 后的阴极双极板吸酸量占总磷酸含量的31.07%,阳极双极板吸酸量占2.07%,而尾气中的磷酸占比不足0.44%;在500h、0.6~1.0A/cm2的负载循环测试中,阴阳两极双极板吸酸占比为21.73%,尾气中的磷酸占比为14.25%,这些数据意味双极板吸酸对磷酸损失以及电池性能衰减的贡献可能被严重低估。
(4)磷酸迁移 在HT-PEMFC 中,磷酸盐物种在电流驱动下存在从阴极向阳极迁移的现象,如图10 所示。这一现象会导致磷酸在膜内形成浓度梯度,而磷酸浓度梯度则为磷酸反向扩散提供了驱动力,最终膜内磷酸在两个驱动力的作用下达到动态平衡,然而受限于聚合物分子链与磷酸间的相互作用,大约只有2%~4%的电流来自于离子迁移[38]。虽然磷酸迁移本身不会造成磷酸的损失,但是迁移过程会影响磷酸的浓度与分布,从而影响到磷酸损失速率。

图10 磷酸迁移示意图[38]
早些年,ABT、EDS、ICP 等方法常被用于非原位的磷酸含量测定,然而这些方法无法反映实际电池运行过程中的磷酸分布,因此一些原位测试手段在近十年被相继开发,例如同步辐射X射线透射成像[14-15]、电子探针显微分析[6,39]、同步辐射中子成像[40]、同步辐射X 射线断层显微镜[41-42]、电子顺磁共振[43]以及原位微电极[38]等。基于这些原位测试手段,研究者们对膜内磷酸迁移机理以及其影响进行了较为细致的研究。
首先,磷酸迁移被认为是一个快速过程,虽然膜内磷酸达到稳态往往需要数小时,但大部分磷酸在前10min就已经完成了迁移,这些迁移的磷酸有可能会引起阳极酸淹,导致氢传质阻力增加以及阴极催化层磷酸浸润度降低[44]。在电池寿命后期,磷酸迁移可能会加剧阴极催化层磷酸的消耗,并且PBI 小分子可能随磷酸迁移并沉积在阳极催化层,从而导致催化剂活性区域的减少[6,39]。此外磷酸分布的不均匀还会引起膜的局部膨胀[15]以及微观电极片段的扭曲倾斜[14],从而导致MEA结构被破坏。
图11 磷酸溶液电导率与磷酸浓度关系[45]


图12 电流密度与迁移关系[41]
此外,磷酸还可能会通过双极板的微孔/微裂纹或密封圈的光滑表面从一个单池转移到另一个单池,这一现象在PAFC 中较为常见,但目前在HTPEMFC领域中还没有详细的报道。
1.3 高温质子交换膜性能衰减的缓解策略
1.3.1 膜材料改性
膜材料的改性主要集中于提高PBI膜的机械强度、储酸能力以及化学稳定性,常见的改性方法如下。
(1)改变制备工艺 该方法主要包括增加聚合物平均线性分子量以及高温热处理。其中聚合物分子量的提高可以有效地提升PBI膜的机械强度与化学稳定性,同时由于机械强度的提高,膜内可以掺杂更多的磷酸,因此该方法也可以改善膜的储酸能力与电导率[46]。而高温处理可以使膜发生热交联,使其拥有高抗溶胀性、高机械强度以及不溶性等热固性树脂的特性,同时该方法也能显著提高膜的化学稳定性[47]。
(2)改变PBI分子链结构 该方法在过去十余年内非常常见,通常的做法是在PBI主链上引入吡啶、对苯二酚、磺化芳基、叔丁基、氟化烷基等基团以改变PBI 膜的性质。例如吡啶基[48]、硫原子或磺酸基[49]的存在通常可以提高膜的化学稳定性、热稳定性、储酸能力以及溶解度;F原子与Si原子能改善膜的力学性能和稳定性[50];叔丁基、乙基以及氟化烷基等基团会增加聚合物的间距,从而提升膜的储酸能力[51]等。
(3)构建支化聚合物 通常含有支化结构的PBI膜具有优秀的储酸能力,通过引入膦化基团可以进一步增强膜内电导率并减少磷酸损失[52],但是传统支化聚合物在高磷酸掺杂水平(ADL)下的机械强度低[53],因此需要结合化学交联[54]等方法来改善膜的力学性能。
(4)取代活性位点 PBI 分子链中端基与苯并咪唑环中的NH基团具有较高的化学活性,因此可以使用烷基、苯基、磺酸基等低活性基团来取代咪唑环上的NH基团[11],或者用苯甲酸[55]、尿素[56]等将端氨基转化为更为稳定的苯并咪唑酮。然而需要注意的是,NH 基团被取代会破坏膜内氢键,导致膜的热稳定性降低,并影响膜内ADL与质子电导率。


图13 化学交联示意图
(6)构建嵌段共聚物 由于嵌段共聚物中不同嵌段间的物理化学性质不同,因此其膜内会形成纳米相分离结构。这种相分离结构不仅有利于质子传输,同时还可以结合不同聚合物间的优点,例如p-PBI[70]可以提高膜的机械强度,m-PBI[70]可以提高其加工性能,聚亚芳基醚砜[71]能够促进质子传导并保持尺寸稳定性,磺化PBI[72]可以改善质子传导等。不同聚合物的比例与嵌段长度均会影响嵌段聚合物膜的物理性质。
(7)引入无机填料 二氧化硅[73]、二氧化钛[74]和氧化钡锆(BaZrO3)[75]等无机氧化物通常可以改善膜的力学性能和质子传导率,然而氧化物在磷酸中的稳定性需要被认真考虑[12]。多壁碳纳米管(MWCNT)可以大幅提升膜的机械强度,并且其表面经过膦化或磺化后可以改善增强膜的质子电导率,例如Nafion 功能化可以形成离子交联结构,PBI功能化可以增强其相容性[76-77]。然而MWCNT具有导电性,因此需要注意电池内部短路的问题。氧化石墨烯(GO)[78]的引入通常可以提高膜的质子电导率、磷酸保留能力以及抗氧化性,其本身为电子绝缘体,经过膦化[78]或磺化[79]后具有质子传导能力。磷钨酸(H3PW12O40)[80]、硅钨酸(H3SiW12O40)[80]、磷钼酸(H3PMo12O40)[81]等杂多酸(HPA),以及磷酸氢锆[Zr(HPO4)2·nH2O][80]、磷酸硼(BPO4)[82]、锡基焦磷酸盐[83]、磺化苯基磷酸铯(CeSPP)[56]等磷酸盐也被报道可以改善膜的质子电导率以及机械强度。
(8)构建基质加强膜 目前报道的基质材料有聚四氟乙烯(PTFE)[84-85]、聚醚醚酮(PEEK)[27]、聚偏二氟乙烯-六氟丙烯(PVDF-HFP)[86]等。该方法可以用来提高膜的机械强度与稳定性,以及减少气体渗透率。其中PTFE 与PBI 之间的化学相容性较差,目前文献中报道了化学活化法[84]和偶联剂预处理法[85]两种方法来改善PTFE 与PBI 间的界面性能。
(9) 添加自由基淬灭剂 该方法常见于PEMFC 领域,通常的做法是在电极或膜内引入过氧化氢分解催化剂或自由基猝灭剂,如Ce、Mn、MnO2、SnO2、CeO2以及对苯二甲酸等[87]。近期Hao等[88]证实了CeO2纳米颗粒能够提高PBI膜的化学稳定性,机理如图14 所示,这意味着该缓解策略有希望应用于HT-PEMFC体系。
1.3.2 磷酸管理策略的改进
HT-PEMFC 中磷酸的损失对电池寿命的影响是不可忽视的,目前磷酸管理的改进策略主要有以下几种。

图14 CeO2消除自由基机理与Ce3+活性位点再生机制[87]
(1)抑制磷酸蒸发 该策略包括在出口处设置没有催化剂的冷凝区以及设置磷酸预饱和装置[89]。其中前者可以将冷凝区的温度降低近30℃,从而使得出口处磷酸蒸汽压降低了4倍。而后者则可以有效地抑制MEA内磷酸蒸汽向流道的扩散。
(2)降低石墨板吸酸程度 该策略包括提高石墨板的疏水性以及减少表面孔隙率,通常的做法是添加疏水黏结剂[37]或进行表面预处理[90],例如Pilinski 等[37]发现在石墨双极板中引入酚醛树脂或CNT均能有效地降低双极板的吸酸量。
(3)抑制酸淹 该策略主要包括减少磷酸迁移路径以及固定质子导体。前者主要通过降低微孔层与催化层的裂纹密度[42]来实现,而后者主要通过引入无机氧化物Al2O3[91]以及CsHSO4[92]、Zr(HPO4)2[93]、磺化苯磷酸铁(FeSPP)[94]、PVPA[95]等固体酸来构建稳定的质子通道,从而起到代替磷酸并抑制酸淹的作用。
(4)增强膜与磷酸的相互作用 该策略包括提高膜内ADL 以及限制膜内磷酸的迁移。通常,能够提高膜机械强度以及质子电导率的膜改性策略均有利于增强膜与磷酸的相互作用,详见第1.3.1节。
(5)抑制磷酸进入气体扩散层 磷酸在高电流密度下进入气体扩散层是磷酸损失的一大途径,目前的研究认为催化层界面与微孔层界面的裂纹网络间的重合点是磷酸进入气体扩散层的主要途径[42,96]。而这些裂纹可能来自于MEA 制备时的电极干燥过程[97],因此减少微孔层的微裂纹可以有效地抑制磷酸进入气体扩散层,减少磷酸流失。
2 气体扩散电极性能衰减机理与缓解策略
2.1 气体扩散电极材料特性
气体扩散电极(GDE)是HT-PEMFC 的关键部件之一,通常由催化层(CL)、微孔层(MPL)和气体扩散层(GDL)组成。
2.1.1 催化层材料特性
催化层是电化学反应的主要场所,主要由催化剂、碳载体以及黏结剂组成,其中催化剂是电化学反应快速进行的关键。目前,HT-PEMFC 阳极催化剂主要为Pt 催化剂,而阴极催化剂主要为Pt 或Pt/Co 催化剂。为了提高Pt 催化剂的利用率,通常会将2~5nm 的Pt 纳米颗粒分散到具有高比表面积、高电导率以及高稳定性的载体表面,目前最为常用的催化剂载体是炭黑,例如Vulcan Carbon 以及Ketjen Blacks,前者表面积为200~300m2/g,而后者为700~1600m2/g[34]。由于后者的高比表面积,其催化剂性能表现更为优异,然而,前者具有更高的稳定性和抗氧化能力,因此更适合用于HTPEMFC[98]。近些年,一些新型碳纳米材料(如碳纳米管[99]、碳纳米球[100]等)也被用作HT-PEMFC催化剂的载体材料,而且表现出更为优异的稳定性。
除了上述的催化剂与碳载体,催化层中还会引入一定量的黏结剂,用来改善气体传输以及提高催化层机械稳定性。目前用于HT-PEMFC 的黏结剂主要分为吸酸型和疏水型,如图15所示。吸酸型黏结剂主要包括m-PBI[101]以及AB-PBI[102],其作用是控制催化剂中的磷酸分布。然而此类黏结剂容易引起气路阻塞或酸淹[103],同时催化层气相通路结构的稳定性也可能存在问题[34]。疏水型黏结剂主要包括聚四氟乙烯(PTFE)[104]和聚偏氟乙烯(PVDF)[105]。相比于吸酸型黏结剂,疏水型黏结剂能够在催化层内形成稳定的气相扩散通路,具有更加优异的电极性能,其中PVDF的分散更为均匀,但其熔点仅为170℃;而PTFE 分散较为困难,但其熔点可达327℃,热稳定性更好。
2.1.2 气体扩散层与微孔层材料特性

图15 HT-PEMFC的催化剂结构示意图[34]
气体扩散层在电极中主要起分配气体、提供电子传输路径以及为催化层提供机械保护的作用。而微孔层主要用来改善催化层与气体扩散层间的界面状态,降低接触电阻以及限制磷酸浸出。
目前HT-PEMFC 中气体扩散层常用的材料为碳纸与碳布,如图16 所示。两种碳纤维材料都具有足够的导电性和化学稳定性,孔隙率通常大于70,具有良好的气体输送能力。其中碳纸是由随机排列的碳纤维丝堆积黏合制得,通常需要黏合剂(例如碳化树脂),密度较高;碳布则是由碳纤维丝经纺织工艺制得,具有高机械弹性、低密度和高渗透性等特点。此外,为了提高碳纸或碳布的疏水性,通常还会引入聚四氟乙烯(PTFE)涂层,其质量分数一般在5%~30%[106]。HT-PEMFC 中的微孔层通常是由亚微米级的石墨颗粒与聚合物黏结剂混合制备而成,经热处理黏合到电极表面,厚度一般为20~50μm[106]。

图16 HT-PEMFC中气体扩散层表面形貌[107]
2.2 气体扩散电极性能衰减机理
目前对于气体扩散电极性能衰减的研究多集中于催化层内,Shao 等[108]和Zhang 等[109]曾对PEMFC中催化层的性能衰减机理进行了详细综述,其主要衰减方式如表3 所示,虽然HT-PEMFC 与PEMFC存在一定差异,但其催化层性能衰减机理相似。
2.2.1 催化剂溶解与脱合金化
铂基催化剂在浓磷酸溶液中存在着溶解平衡,该过程通常包括电化学溶解与化学溶解。其中Pt的溶解度随电势的升高呈指数上升,且随温度的升高而少量增加[110]。对于纳米级的Pt 颗粒而言,Pt更容易通过直接的电氧化反应形成Pt2+[111],如式(7)所示,随着电极电位的升高,Pt 的电氧化过程加剧,同时PtO的生成均会导致其化学和电化学溶解速率显著提升[112],如式(8)和式(9)所示。在HTPEMFC中,阳极侧处于低电位的还原气氛,而阴极侧则处于高电位的氧化气氛中,因此通常认为催化剂的溶解主要发生在阴极侧[113]。然而,由于较高的环境温度与铂纳米颗粒较低的溶解电位,HTPEMFC阳极催化剂的溶解可能被严重低估[97]。

表3 催化层性能衰减机理

另外,在PtCo/C、Pt3Ni/C 以及PtRu/C 等铂合金催化剂中,催化剂的合金化程度以及掺杂的金属种类被认为是影响其在酸性环境中稳定性的关键因素,其中Pt/Cr 或Pt/Co 合金显示出较高的稳定性[114],而Pt/Ru催化剂的稳定性较差,其掺杂的Ru更容易从催化剂颗粒中脱离[115]。在HT-PEMFC中,由于温度的提升,非铂金属元素的脱合金化现象更加严重[97]。

图17 铂迁移示意图[97]
2.2.2 催化剂迁移与铂带的形成溶解的铂离子在MEA 内部存在迁移现象,如图17 所示。一般来说,阴极溶解的铂离子主要通过浓差扩散[116]以及电渗拖曳[117]进行迁移,而阳极溶解的铂离子主要通过浓差扩散以及电迁移作用[118]进行迁移。由于阴极催化剂溶解更为严重,因此整体表现出催化剂从阴极向阳极迁移的趋势。其中部分游离的催化剂离子在催化层与膜的界面附近被氢还原,发生沉积并最终形成铂带[119],如图18 所示。这一现象在PEMFC 以及PAFC 中被广泛报道[21,120],其中铂带的位置与膜内氢气和氧气的相对分压有关[121],而铂带的形状则与形成时间有关[118]。通常铂带的形成会使得H2与O2在此处迅速反应,从而出现局部热点,同时该反应可能会促进H2O2生成,加速膜的化学降解。

图18 MEA截面SEM图[119]
近期在HT-PEMFC体系中,Liu等[116]利用SEM/EDS在活化70h的MEA中发现了铂带的形成,其形成规律与PEMFC 中的相似;Hengge 等[97]利用SEMTEM同样在阳极(采用Pt/Ru催化剂)与膜的界面处观察到了明显的催化剂颗粒带,此外利用EDX 在靠近阳极的膜内观测到大量以Ru 为主的催化剂颗粒,这一现象被认为是阳极催化剂溶解迁移的直观证据。
2.2.3 催化剂粒径增加
催化剂平均粒径的增加是电池运行过程中ECSA 降低的主要原因,这一现象通常会导致电池性能的持续衰减。在PEMFC 的研究中,铂颗粒的粒径增加机制主要有以下2种。
(1)Ostwald 熟化机制 在该机制中,溶解后的铂离子在最低表面能的驱动下,会倾向于沉积在较大的催化剂颗粒上,从而导致铂颗粒粒径逐渐增加,通常这一过程会导致粒径分布曲线整体右移,同时分布更为集中[122]。此外,溶解后的铂离子也可能以新晶粒的形式在离聚物相中或是碳载体表面析出,但是该过程可能会产生更为细小的铂颗粒,因此其粒径分布会发生宽化[21]。
(2)催化剂颗粒团聚 催化剂颗粒可能会从碳载体上脱落,而这些催化剂颗粒经过迁移、烧结,会逐步形成粒径较大的团聚体[123-124]。这一过程通常来自于催化剂颗粒间的随机碰撞,因此其粒径分布呈现出典型的对数正态分布[124]。
在HT-PEMFC 中,催化剂粒径增加的机理与PEMFC中的相似,但由于其特有的酸性高温环境,上述机制均会被加剧。通常催化剂粒径增加主要发生在HT-PEMFC寿命初期[6],期间催化剂颗粒的团聚被认为是铂颗粒粒径增加的主要途径[124],并且阴极侧催化剂粒径往往远大于阳极侧[113,125]。这些现象可以理解为在电池寿命初期,碳载体表面的铂颗粒平均粒径小、分布广,高温高电位下更容易发生碳腐蚀与铂的迁移团聚,然而在电池寿命后期,平均粒径的增加限制了上述过程,从而缓解了由催化剂团聚引起的性能衰减[9]。
2.2.4 碳腐蚀
在HT-PEMFC 中,碳载体的腐蚀往往是导致催化层发生降解的一个重要因素,通常该过程会削弱铂颗粒在碳载体上的附着,从而加剧铂颗粒的脱落与团聚[108],同时长期碳腐蚀过程还会降低电极的疏水性并破坏电极结构的完整性,这可能会导致传质阻力与磷酸损失增加[34]以及电极变薄。
在酸性溶液中,纯碳与水直接反应生成二氧化碳的平衡电势仅为0.207V(vs. SHE),如式(10)所示[126],这意味着电池运行过程中,阴极碳腐蚀在热力学上是可行的,但是由于该反应的动力学过程极为缓慢,因此低电位时的碳腐蚀速率极低。然而碳腐蚀速率会随着电位的升高呈指数上升[127],在动态操作过程中或开路状态下,阴极会暂时处于高电位状态,甚至局部界面电势差会达到1.4~1.6V,此时的碳腐蚀速率会大大增加,并且在HT-PEMFC中,操作温度的提升会进一步恶化这一过程[42]。

此外,催化剂的存在会大大加剧碳腐蚀过程。对于纯碳电极,通常在电极电位达到0.9V(vs.SHE)以上时,才能够观察到明显的碳腐蚀现象,而掺杂铂催化剂的碳电极则可以在0.6~0.9V(vs. SHE)区间观察到明显的电化学碳腐蚀[128]。目前的研究认为,碳载体通过表面氧化与催化氧化形成CO2是电极发生电化学碳腐蚀的主要反应途径[129],如式(11)和式(12)所示。在这一过程中,碳表面会生成含有醚键、羰基、羧基、内酯或酚基等基团的碳氧化物种[130-131],随着电位的增加,这些碳表面氧化物会在催化剂的作用下发生电化学反应,因此催化剂周围的碳更容易发生电化学腐蚀,铂颗粒更容易从碳载体上脱落并发生团聚。

除了上面提到的电化学腐蚀,化学腐蚀也有可能对整体碳腐蚀有所贡献。Willsau 等[128]曾报道称H2O2的存在会导致整体碳化学腐蚀增加。而近期Engl等[132]则提出了一个基于碳表面活性位点的化学氧化机理,如式(13)和式(14)所示,这一机理被用来解释启停循环过程中的阳极碳腐蚀的问题。

2.3 气体扩散电极材料改进策略
催化剂溶解团聚和碳腐蚀是造成电池性能持续衰减的主要原因,目前HT-PEMFC 中的电极改进策略主要有以下几种。
(1)提高碳载体的耐腐蚀性 提高碳载体的石墨化程度是增强碳载体耐腐蚀性的常见策略,例如高温处理炭黑[133]或采用碳纳米材料[98,129,134],其中提高炭黑的石墨化程度是最为简易的方法,然而该策略会牺牲部分表面积[98,129]。
(2)提高催化剂结构的稳定性 对于铂基催化剂,构建特殊的催化剂结构也是一种设计思路。在PEMFC中,Lin等[135]构建了具有核壳结构的Co@Pt/C催化剂,这种结构不仅有利于提高催化剂的稳定性,同时Pt对Co的包覆也抑制了Co的溶解。Wang等[136]构建了Pd@Pt3Co/C 核壳结构催化剂,同样表现出优异的性能与稳定性。在HT-PEMFC体系中,You 等[137]利用多元醇法制备了PdNiCu@PdIr/C 核壳结构催化剂,在4000h 的稳态测试中,其MEA 性能衰减速率仅为12.4μV/h,表现出较好的ORR 活性与稳定性。此外,一些研究者通过在WMCNT或多孔纳米碳颗粒[138]上涂覆PBI[99]或PyPBI[139]来构建稳定的催化剂/载体结构,如图19所示,其实验结果均显示出较高的耐腐蚀性。

图19 CNT/Pt/PBI电极结构示意图[99]
(3)增强载体与催化剂的相互作用 该策略通常的做法是在碳载体的基础上引入过渡金属氧化物,例如TiO2[140]和ZrO2[141],通过抑制催化剂的生长与团聚来提高催化剂的稳定性,同时该策略还有利于改善催化剂的活性[120]。
(4)提高电极疏水性 H2O被认为是碳腐蚀过程中氧的主要来源,水含量的增加会明显提升电极碳腐蚀的速率[142]。在PEMFC 中,Ferreira-Aparicio等[143]曾利用电喷雾法制备了一种超疏水电极,其在启停衰减测试中表现出较好的耐腐蚀性,然而目前HT-PEMFC 领域内还没有相关报道,该策略的适用性还有待考察。
(5)替换载体材料 目前,硼掺杂金刚石[144]、碳化钨微球[145]、SiC[146]、氮化钛纳米颗粒[147]、TiO2[148]、二氧化钨(WO3-x)[149]以及碳化硅钛(SiCTiC)[150]等载体材料在HT-PEMFC 中均有报道。然而相比传统的碳材料,虽然这些材料拥有更加优异的稳定性与力学性质,但同时也存在表面积低、分散困难或导电性能差等问题,因此目前该策略仍需进一步地探索。
3 双极板性能衰减机理与缓解策略
3.1 双极板材料特性
双极板是燃料电池中的关键部件之一,它在电池内主要起到机械支撑、物料分配、热量传递以及电子传导的作用。目前HT-PEMFC 中的双极板主要分为石墨双极板、复合双极板以及金属双极板三类。
早期HT-PEMFC 中通常采用的是石墨双极板,其本身在高温酸性环境中具有优异的化学稳定性与导电性。然而这种石墨双极板孔隙率高、机械强度低,加工困难。为了改善这些缺点,一些研究者基于石墨和聚合物(例如聚丙烯[151]、聚苯硫醚[152]、酚醛树脂[152]、乙烯基酯树脂[153]、环氧树脂[154]等)开发了石墨复合双极板。相比传统的纯石墨板,这种复合双极板重量更轻、力学性能更好、更易于加工,但聚合物的加入牺牲了部分导电性能,导致电池输出功率降低。相比石墨/复合双极板,金属双极板机械强度极高,其厚度一般为0.1~0.2mm,同时金属双极板还具有高导电性、导热性以及气密性等优点。然而金属双极板往往面临着严重的腐蚀问题,尤其是在HT-PEMFC 体系中,高温酸性环境进一步加剧了金属的腐蚀程度,因此目前在HTPEMFC领域中关于金属双极板的报道较少。
3.2 双极板腐蚀特性
3.2.1 石墨/复合双极板
石墨/复合双极板在HT-PEMFC 中的稳定性相对较好,除了之前提到的双极板吸酸问题[35],目前关于石墨/复合双极板在HT-PEMFC 中腐蚀情况的研究[155]并不多。相对而言,由应力变化引起的机械变形以及聚合物高温热解对石墨/复合双极板稳定性的影响可能更为严重,因此目前的文献报道主要集中于改善其力学性能、导电性能以及热稳定性[155-156],这里不再进行过多的阐述。
3.2.2 金属双极板
金属双极板在热磷酸中的腐蚀特性在近20 年内受到了广泛关注,研究者们测试了奥氏体不锈钢(例如201、304、316L、317L、904L 等)、镍合金(例如Cronix80、HastelloyC-276 等)、Ti、Ta、Nb以及SiC等基体材料在高温浓磷酸溶液中的耐腐蚀性[157-162]。其测试结果显示这些材料的腐蚀速率与测试温度以及酸浓度呈正相关,并且随电极电位的改变而改变。在150℃、模拟阴极电位条件下,不锈钢的腐蚀速率约为1mm/a,其表面氧化层主要为氧化铬[158,162];镍钼与镍铬合金的腐蚀速率约为0.03~0.04mm/a,其中镍铬合金耐腐蚀性能最好[159,162],而镍钼合金在阳极极化条件下稳定性较差[159,162];热压SiC 的腐蚀速率约为0.1mm/a,同样,其在阳极极化条件下不稳定[162];Ti 和Nb 在高温环境下均表现出较差的稳定性[160-161];Ta 金属单质的腐蚀速率仅为1μm/a,并且无论是作为涂层还是合金组分,其都能显著提高基体的耐腐蚀性[159-162],然而钽的成本远远高于不锈钢与镍合金。
单从上述结果来看,似乎没有合适的基体材料。然而需要注意的是,这些实验是在浓磷酸溶液中进行的,在实际运行过程中,其腐蚀情况可能并没有想象中的那么严重。近期,Alnegren等[157]设计的SS316L 金属单电池在900h 内的电压衰减仅为19μV/h,而Janßen 等[163]设计的镀金SS316L 金属电堆(5 节)在前2000h 的电压衰减速率为28μV/h,最终寿命约为4000h,如图20所示。这些测试结果意味着不锈钢、镍合金等价格低廉的金属材料仍然有希望应于用HT-PEMFC。

图20 德国于利希研究中心SS316L镀金双极板电堆寿命测试曲线[163](160℃,氢气/空气化学计量比为2/2)
3.3 金属双极板腐蚀缓解策略
目前的研究认为表面涂层是保证金属双极板长期使用的关键[157]。在HT-PEMFC 中,早些年研究者曾用金作为金属表面涂层[35,164],然而镀金的成本很高,Janßen 等[163]报道的镀金双极板金含量高达0.45g/片,这显然不利于其商业化。近些年,钽由于其极为优秀的耐腐蚀性与相对可接受的成本被认为是HT-PEMFC 金属双极板的理想涂层。Christensen等[162]的测试结果显示,钛合金表面钽涂层在150℃浓磷酸中的腐蚀速率仅为4μm/a。然而钽表面五氧化二钽钝化膜的形成会大大增加其接触电阻,为了解决这一问题,Wang等[165]借鉴了PEMFC中的思路[166],在Ta/SS430 表面引入了具有高化学稳定性与导电性的氮化钽,其结果显示在0.6V(vs.OCV)时,双极板腐蚀电流仅为0.93μA/cm2,而接触电阻仅为9.03mΩ·cm2。
此外,常见的表面处理技术,例如物理气相沉淀(PVD),会不可避免地引入微裂纹或微孔[165],这些缺陷可能会导致电解液直接接触基体材料,从而加剧局部腐蚀,甚至导致涂层脱落。为了避免这种情况,常用的策略是涂覆多层涂层[166-167]。该策略能够有效地降低孔隙的出现几率并抑制孔隙的进一步腐蚀,同时该策略可以通过选择合适的涂层组合来强化涂层的耐腐蚀能力。
4 密封件能衰减机理与缓解策略
4.1 密封件材料特性
密封件在HT-PEMFC 中主要起密封、补偿公差、电绝缘等作用。橡胶与塑料是最常见的两类密封材料[34],包括硅橡胶、四丙氟橡胶、全氟橡胶、氟硅橡胶、乙丙橡胶、聚四氟乙烯、全氟乙烯丙烯共聚物等[9]。其中硬质密封件具有较高的机械强度,容易控制厚度与形变程度,但缺点是密封可靠性差,对公差的补偿能力差;弹性密封件在机械稳定性方面更占优势,但其压缩比难以控制[34]。两类密封件各有优缺点,在目前的HT-PEMFC 中均有应用。
4.2 密封件失效机理与缓解策略
在HT-PEMFC 中,密封件失效往往会引起气体或冷却剂泄露,且密封件本身的降解产物也可能会污染膜电极,这些情况均会导致电极性能的持续衰退。目前由HT-PEMFC 密封件失效引起电池性能衰减的报道很少,但根据PEMFC 领域的相关报道以及高分子材料特性,可以推测其失效方式可能有以下几种。
(1)机械降解 在高温高压条件下,高分子材料往往会发生蠕变、硬化以及应力松弛等现象,通常这些过程比较缓慢,但在电池寿命后期,这些现象可能会导致密封件难以提供足够的压力来维持气密性。此外压力分布不均匀、压力过高或频繁的应力变化均会加速高分子材料的断裂;不合理的密封间隙则可能导致密封件局部变形破损。对于硬质密封件,由于压力波动或震动,其与双极板之间可能还会存在细微的相对运动,这会引起密封件表面磨损,从而导致密封性能变差[168]。
(2)热氧化降解 在氧气环境中,由于过氧化物与氧化自由基的生成,高分子材料会发生自催化的热氧化降解,该过程的降解温度远低于惰性气氛下的降解温度,同时降解速率更高。虽然在筛选阶段会尽可能选用热稳定性高的密封材料,但在局部高温热点处、异常升温或长时间运行情况下,密封件仍然可能面临热氧化降解的问题。
(3)化学降解 在PEMFC 中,密封件(例如硅橡胶)的化学降解通常会导致其表面裂纹扩张以及光滑度降低,其降解机理包括高分子主链的断裂和交联位点的水解,其中酸[169-170]与水[171]的存在均会加剧其降解程度。在HT-PEMFC 中,由于热浓磷酸的存在,密封件的化学降解问题可能会更加严重。此外,聚合物小分子还可能会在热的浓磷酸中溶出,进而导致密封件密封可靠性的降低以及膜电极的污染。
目前HT-PEMFC 领域并几乎没有关于密封件失效缓解策略的详细报道,但随着膜电极组件与双极板研究的不断完善,新型密封材料的开发以及密封结构的设计可能会受到更多关注。
5 结语
HT-PEMFC 在电池材料耐久性问题上面临的挑战要比PEMFC 严峻得多,其中导致电池性能衰减的主要原因包括膜降解、碳腐蚀、催化剂溶解团聚、双极板吸酸/腐蚀以及密封件失效等。
虽然目前材料的耐久性问题已经得到很大的缓解,但一些工程问题仍然制约了HT-PEMFC 系统的寿命,例如石墨复合双极板机械强度不足、体积大,常见的密封件材料在高温酸性环境中稳定性不足,以及膜电极贵金属载量高、成本高等问题。这些工程问题给HT-PEMFC 电堆的设计、系统集成以及未来的商业推广都带来了很多麻烦,因此未来耐腐蚀性金属双极板和高稳定性密封件材料的开发、催化剂载量优化以及廉价材料的替换等研究方向有望成为未来该领域内的研究重点。
庆幸的是,虽然HT-PEMFC 距离大规模应用还有很长的路要走,但是其在商业领域已经积累了一定的技术基础,例如丹麦的Danish Power Systems、SerEnergy,美国的Advent Technologies 等公司都已经推出了基于HT-PEMFC的实用化商品。此外,一些高校与研究机构,例如丹麦科技大学、奥尔堡大学、德国于利希研究中心、中国科学院大连化学物理研究所等,也均参与着HT-PEMFC 的改进工作,这意味着未来的HT-PEMFC 市场将充满着竞争与活力。
