气相色谱法测定土壤中石油烃含量的不确定度评定
2020-06-28周吉峙段路路
张 吉,周吉峙,段路路
(1.上海大学环境与化学工程学院 上海 200444;2.上海化工研究院有限公司 上海 200062)
试验数据的准确性和可靠性事关实验室的核心竞争力,也是实验室资质认证的关键。然而,由于真值的未知性和不确定性,而误差又是客观存在且不可避免,测量的不确定度也由此应运而生。测量的不确定度是测量结果质量的重要指标,是目前对于误差分析的最新理解和阐述,它表示由于测量误差的存在而对被测量值不能确定的程度。
由于经济和社会的发展,土壤、水体和空气作为人类赖以生存的物质基础已经受到不同程度的污染。特别是进入现代社会后,原油的开采以及石油产品的广泛应用,造成了土壤中大量的石油烃污染。目前,检测土壤中石油烃含量的方法比较成熟,主要有重量法、红外光度法、紫外分光光度法、荧光分光光度法和气相色谱法,其中气相色谱法可以同时对多个组分进行定性定量分析,且能获得样品的特征色谱数据,目前在国内外都有逐渐取代其他方法的趋势[1]。因此,对气相色谱法测定土壤中石油烃含量的不确定度评定显得尤为必要。为此,根据《测量不确定度评定与表示》(JJF 1059.1—2012)[2]和《化学分析中不确定度的评估指南》(CNAS-GL06)[3]并参照EPA Method 8015c(美国国家环保局方法 8015c)[4],采用气相色谱法对土壤中石油烃的含量进行测定,并建立起针对气相色谱法测定土壤中石油烃含量的较为完整的不确定度评价方案。
1 测定用仪器及测定步骤
1.1 主要仪器和试剂
主要仪器:7890A气相色谱仪,美国安捷伦科技有限公司;AL204-1C分析天平,220 g/0.1 mg,e=1 mg,瑞士梅特勒公司;PPM48半自动正压固相萃取仪,美国J2Scientific公司;E-916快速溶剂萃取仪,瑞士步琦有限公司;平行蒸发仪,瑞士步琦有限公司;20~200 μL、100~1 000 μL、1~10 mL移液器,德国普兰德公司。
主要试剂:二氯甲烷,色谱纯,CNW;正己烷,色谱纯,CNW;无水硫酸钠,分析纯,国药集团化学试剂有限公司;固相萃取柱子,CNWBOND Si SPE Cartridge 500 mg 6 mL;石油烃标准物质,o2si n-Alkane Solution,C7to C40,1 000 mg/L,1 mL。
1.2 仪器工作参数及环境条件
(1)工作参数
色谱柱:Agilent 19091 J-413,325 ℃,30 m×20 μm×0.25 μm,HP-5。
前置进样口:不分流进样;加热器:300 ℃;压力:46.223 kPa(6.704 1 psi);总流量:19.5 mL/min;隔垫吹扫流量:3 mL/min;隔垫吹扫流量模式:标准;载气节省打开:15 mL/min;开始等待时间:10 min;分流出口吹扫流量:15 mL/min,1 min。
前置FID,加热器:320 ℃;H2流量:40 mL/min;空气流量:400 mL/min;尾吹流量(N2):28.5 mL/min;恒定柱流尾吹:30 mL/min;火焰:打开;电位计:打开。前部信号(FID)数据采集频率/最小峰宽:20 Hz/0.01 min。
柱箱运行过程:40 ℃保持5 min,升温至60 ℃保持1 min,再从60 ℃升温至320 ℃保持9 min,整个过程升温速率均为15 ℃/min。
(2)环境条件
温度:(20±5)℃;湿度:50%~70%。
1.3 测定步骤
参照EPA Method 8015c,称取固体样品20 g(实际称样量精确至0.000 1 g),加入50 mL混合溶剂萃取液(V二氯甲烷∶V正己烷=1∶1),放置于快速溶剂萃取仪中进行萃取,加入过量无水硫酸钠干燥,过滤,转移至锥形瓶中;用半自动正压固相萃取仪进行固相萃取,随后用平行蒸发仪进行浓缩,浓缩液用正己烷定容至1 mL,空白试样进行同样的处理;样品溶液采用气相色谱仪进行测定,同时进行空白试样的测定,测定流程如图1所示。气相色谱仪会自动将空白溶液中石油烃的含量进行扣减,得到的测定结果即为样品溶液中石油烃的实测含量。

图1 土壤样品中石油烃含量测定流程
2 测量模型及不确定度来源
2.1 测量模型的建立
测量模型如式(1)所示:
(1)
式中:W——样品中被测石油烃的含量,mg/kg;
C——样品溶液中被测石油烃的实测质量浓度,mg/L;
V——样品浓缩液的定容体积,mL;
f——样品溶液的稀释因子;
Q——气相色谱仪定量校准影响因子;
m——样品称样量,g。
样品溶液中被测石油烃的实测质量浓度按式(2)计算:
C=Ci-C0
(2)
式中:Ci——样品溶液中被测石油烃的质量浓度,mg/L;
C0——空白溶液中被测石油烃的质量浓度,mg/L。
2.2 不确定度主要来源及分析
由测量模型可知,输出量是各输入量的乘积。因此,根据测量不确定度传播律,样品中被测元素含量的相对合成标准不确定度可写成各输入量的相对标准不确定度的方和根值。则样品中被测元素含量测定的相对合成标准不确定度ur(W)为:
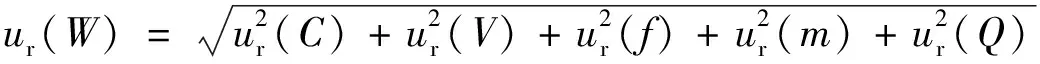
(3)
式中:ur(C)——样品溶液中被测元素实测质量浓度的相对标准不确定度;
ur(V)——样品溶液定容体积的相对标准不确定度;
ur(f)——样品溶液稀释因子的相对标准不确定度;
ur(m)——样品称量的相对标准不确定度;
ur(Q)——气相色谱定量校准引入的相对标准不确定度。
测量不确定度主要来源于如下因素:
(1)标准物质
包括配制标准储备液的不确定度,温度变化造成移液、定容体积变化的不确定度以及稀释过程中引入的不确定度。
(2)样品处理过程
包括选样代表性、样品本身的均匀性,分析天平称量过程引入的不确定度,消解过程的回收率,温度变化对定容体积及移液体积的影响。
(3)样品测定过程的重复性
平行样品测量过程中引入的不确定度包括分析天平称量的重复性、移液体积及定容体积的重复性、萃取回收率的重复性等。
(4)最小二乘法拟合标准曲线
被测元素的测定结果是用标准溶液通过最小二乘法拟合成标准曲线后获得,故须考虑由标准曲线的非线性以及仪器的稳定性等引入的不确定度。
综上所述,参照EPA Method 8015c,采用气相色谱法对土壤中石油烃含量进行测定的不确定度因果关系如图2所示。

图2 气相色谱法测定土壤中石油烃含量不确定度因果关系
3 不确定度的分量评定
3.1 样品溶液中被测元素实测质量浓度的标准不确定度u(C)
样品溶液中被测元素实测质量浓度的标准不确定度u(C)包括标准物质本身不确定度、标准溶液配制过程以及标准曲线拟合时引入的不确定度。
3.1.1 标准物质本身不确定度对质量浓度C测定的不确定度分量u1(C)
根据标准物质附件资料,质量浓度(1 000±50)mg/L,取均匀分布,则:
=0.000 6%
3.1.2 标准溶液配制过程带来对质量浓度C测定的不确定度分量u2(C)
将石油烃的标准储备液(1 000 mg/L)分别稀释配制得到1.25、5.00、12.50、25.00、50.00和100.00 mg/L 6个浓度点的标准溶液,即石油烃(C9~C16)10.00、40.00、100.00、200.00、400.00和800.00 mg/L 6个浓度点的标准溶液和石油烃(C17~C40)30.00、120.00、300.00、600.00、1 200.00和2 400.00 mg/L 6个浓度点的标准溶液。
根据测定不确定度评定的相关规定[3],标准溶液稀释过程不确定度相对于用于拟合直线求实测质量浓度C时的不确定度完全可以忽略不计。采用最小二乘法拟合曲线程序的前提是假定横坐标标准值xi的不确定度远小于纵坐标响应值yi的不确定度,因此,通常实测质量浓度C的不确定度计算程序仅与仪器响应的不确定度有关,而与校准溶液不确定度无关,也不与从同一溶液中逐次稀释产生必然的相关性。所以,u2(C)≈0,u2 r(C)≈0。
3.1.3 标准曲线拟合时引入的不确定度分量u3(C)
试验采用7个浓度水平点(包括空白)的石油烃标准物质溶液,用GC-FID法测定其发射强度,每一浓度水平测定3次,测定结果(表1)用最小二乘法拟合得到直线方程:石油烃C9~C16为y=23.194x-65.323,相关系数r2=0.998 1;石油烃C17~C40为y=17.732x+3 083.7,相关系数r2=0.995 7。则拟合直线方程的标准差u(y)可由式(4)得出:
(4)
式中:yi——xi的仪器响应值;
yfi——xi对应于拟合直线方程求算出的值。
针对实测质量浓度C,由拟合直线引入的标准不确定度u3(C)如式(5)所示:
(5)
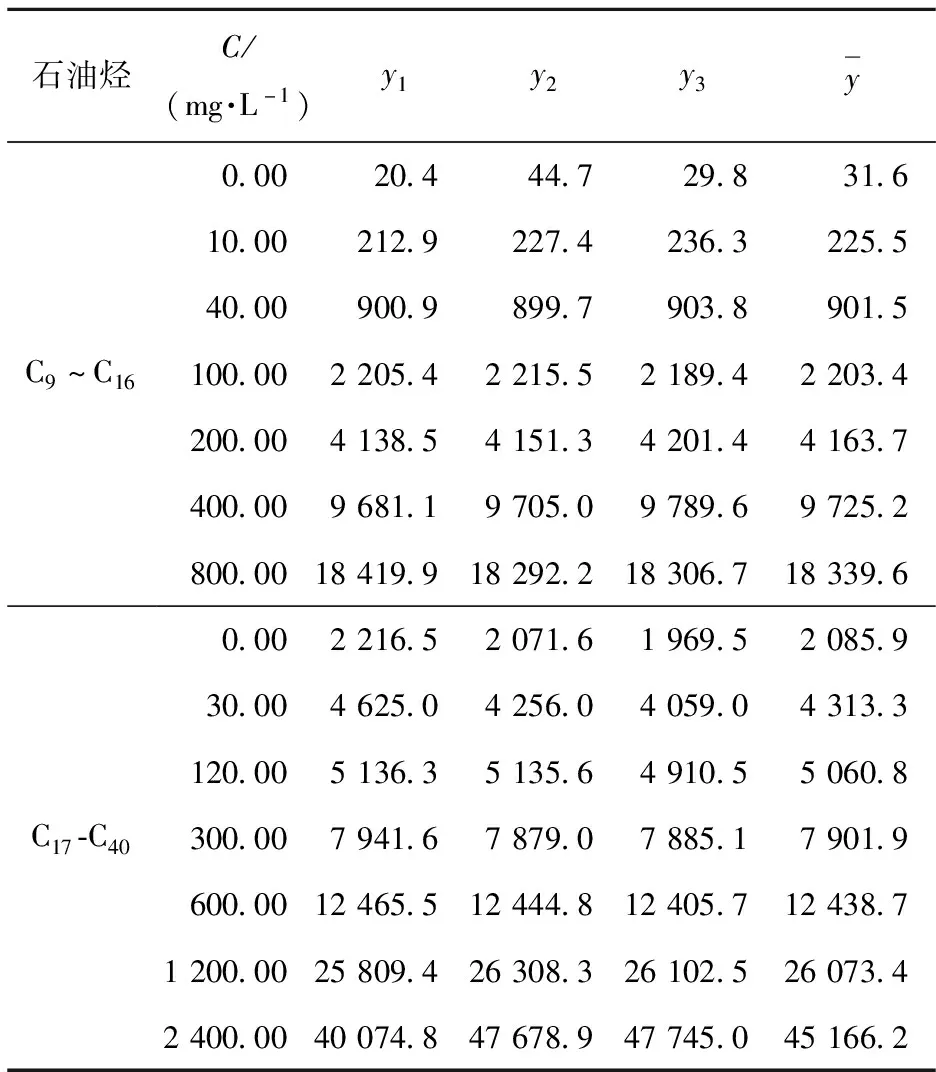
表1 石油烃标准溶液质量浓度与信号面积
式中:b——拟合直线的斜率;
p——试样平行测试次数,p=10;
n——配制7个标准溶液浓度点,每个浓度点测试3次,共计21次,即n=21;
u(y)——工作曲线的标准偏差;

Cp——试验样品测定值,mg/L。
则样品测定值的相对标准不确定度ur(C)=u3(C)/Cp。石油烃的标准不确定度u3(C)及相对标准不确定度u3 r(C)如表2所示。

表2 石油烃的标准不确定度u3(C)及相对标准不确定度u3r(C)
3.1.4 各种随机因素导致的测量重复性引入的不确定度分量u4(C)
在样品快速溶剂萃取过程中存在许多随机因素,包括加入试剂的量、萃取过程的回收率、容器附着的量、移液过程、容量瓶定容过程等,由此导致的样品测定重复性引入的不确定度分量可以通过平行测试样品用A类评定方法评定。在重复性条件下,对试验样品进行10次独立测定,测定结果如表3所示。
表3 试验样品中石油烃含量重复测定结果 mg/kg
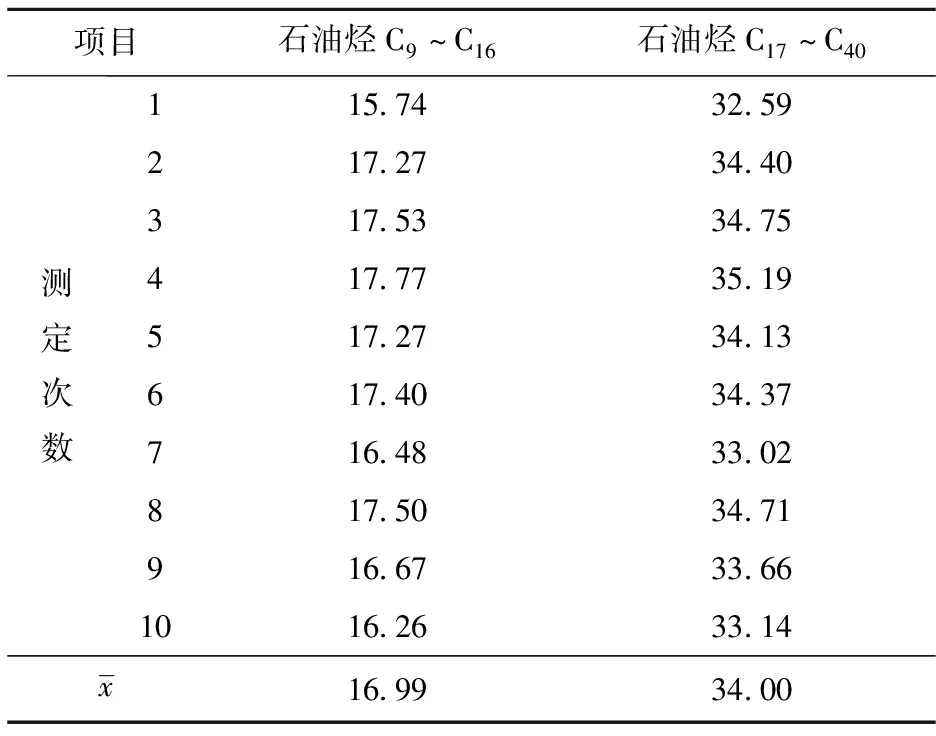
项目石油烃C9~C16石油烃C17~C40测定次数115.7432.59217.2734.40317.5334.75417.7735.19517.2734.13617.4034.37716.4833.02817.5034.71916.6733.661016.2633.14x16.9934.00
经由Grubbs检验[5-6]确认所有10组数据均有效后,分别按式(6)计算算术平均值:
(6)
单次测量的试验标准偏差按式(7)计算:
(7)
以10次测定浓度平均值的标准偏差作为重复性系数的不确定度,按式(8)计算:
(8)
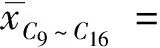
3.1.5 计算样品实测浓度的相对标准不确定度ur(C)
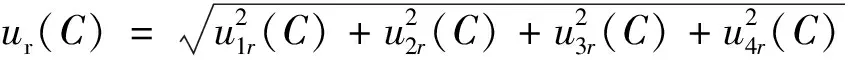
3.2 样品浓缩液定容体积的标准不确定度u(V)
定容过程中的不确定度包括由容量瓶准确性引入的不确定度、由移液器准确性引入的不确定度、读数的重复性引入的不确定度以及温度变化引入的不确定度组成,其中读数的重复性引入的不确定度分量已包含在各种随机因素造成的重复性引入的不确定度分量中。
3.2.1 容量瓶的允许误差引入的体积不确定度分量u1(V)
根据《常用玻璃量器检定规程》(JJG 196—2006)[7],1 mL容量瓶在20 ℃时的容量允许差为±0.010 mL,取均匀分布,则由此引入的容量瓶体积不确定度分量为:
3.2.2 移液器的允许误差引入的体积不确定度分量u2(V)
试验中所用移液器经计量检定合格,根据检定证书,10 mL移液器给出的测量结果扩展不确定度为U=0.004 mL(k=2),由此引入的标准不确定度分量为:
3.2.3 温度变化引入的不确定度分量u3(V)
已知正己烷的膨胀系数α=1.36×10-3℃-1,玻璃器具出厂校准温度为20 ℃,试验使用温度与校准温度变化范围为±5 ℃,按正态分布计算,95%置信概率时k=1.96,所以标准不确定度分量为:

3.2.4 计算定容引入的标准不确定度u(V)
=0.008 1(mL)
其相对标准不确定度为:
3.3 稀释因子f引入的不确定度
对于样品稀释因子,已将其并入样品浓缩液定容体积的标准不确定度,因此不再考虑其对于不确定度的影响,稀释因子对于不确定度的贡献率为0,即u(f)=0、ur(f)=0。
3.4 称量过程引入的不确定度u(m)
称量过程的不确定度主要来自天平的准确性和称量过程的重复性,其中称量过程的重复性引入的不确定度分量已包含在各种随机因素造成的样品测量重复性中。
3.4.1 分析天平的准确性引入的标准不确定度分量u1(m)
试验中所使用的分析天平最大允许误差为0.1 mg,经计量校准合格,其校准证书给出的扩展不确定度为U=0.2 mg(k=2),则:
3.4.2 天平的分辨力引入的标准不确定度分量u2(m)
分析天平的检定证书给出的分辨力为0.1 mg,按均匀分布处理,由此引入的标准不确定度分量为:
3.4.3 计算称量过程引入的标准不确定度u(m)
=0.104(mg)
则由天平称量引入的相对标准不确定度分量为:
3.5 气相色谱仪校准引入的不确定度
根据仪器的校准证书,u(Q)=3%(k=2),则:
4 合成相对标准不确定度
根据式(3),石油烃C9~C16和石油烃C17~C40的相对标准不确定度为:


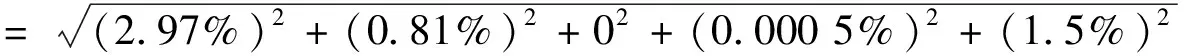
石油烃C9~C16和石油烃C17~C40测定过程中各不确定度分量对合成相对标准不确定度ur(W)的贡献如图3所示。

图3 石油烃C9~C16和石油烃C17~C40测定过程中各不确定度分量对合成相对标准不确定度ur(W)的贡献
由图3结合上述分析计算可见:对于石油烃的测定而言,不确定度的主要影响因素来源于样品浓度测定中标准曲线的拟合和仪器本身的定量校准;另外,定容过程对不确定度带来的影响也不可忽略。
5 扩展不确定度
根据JJF 1059.1—2012[2]的规定,测量不确定度最多取2位有效数字。在95%的置信区间,取包含因子k=2,石油烃C9~C16和石油烃C17~C40的相对扩展不确定度为:
Ur(C9~C16)=urC9~C16(W)×k
=2.38%×2=4.8%
Ur(C17~C40)=urC17~C40(W)×k
=3.42%×2=6.8%
6 结果与讨论
通过建立气相色谱法测定土壤中石油烃含量不确定度评价模型,从而保证对土壤中石油烃(C9~C16和C17~C40)含量测定结果的有效性和合理性,为气相色谱法测定土壤中石油烃含量的质量控制提供了有效、可靠的测量体系。
土壤中石油烃C9~C16的含量可表示为17.0×(1±4.8%)mg/kg,k=2;土壤中石油烃C17~C40含量可表示为34.0×(1±6.8%)mg/kg,k=2。
气相色谱法测定土壤中石油烃的方法有效、可靠、合理、灵敏度高,可以应用于对土壤中石油烃的定性和定量检测。
结果表明,对于石油烃的气相色谱测定而言,不确定度的主要影响因素来源于样品浓度测定中标准曲线的拟合和仪器本身的定量校准;另外,定容过程对不确定度带来的影响也不可忽略。因此,增加标准曲线的浓度水平数以减少由拟合曲线带来的影响,定期对气相色谱仪进行定量校准和及时的阶段性核查显得十分必要;另外,加强对定容仪器的管理和提高实验室分析人员的定容操作能力,有助于减轻定容过程对试验结果的影响。
