213例感音神经性聋患儿的病因分析
2020-06-23戴继任徐彬郑静林乐希刘佳付勇
戴继任徐彬郑静林乐希刘佳付勇*
1浙江大学医学院附属儿童医院耳鼻咽喉头颈外科(杭州310000)
2浙江大学医学院附属儿童医院遗传代谢科(杭州310000)
耳聋是影响人类健康的常见原因。据世界卫生组织统计,全球共有耳聋患者3.6亿,其中儿童占8.9%。我国残疾人群有8296万,其中7岁以下的聋哑儿童数量高达80万,每年新增约11万聋儿[1]。据统计,重度听力丧失的新生儿约为0.1%,其中一半属遗传性耳聋。另外还发现许多迟发性感音神经性听力下降患者的发病是由于自身的基因缺陷或因基因缺陷和多态性造成了对致聋环境因素易感性的增加而致病[2]。因此,遗传性耳聋的分子病因学研究非常重要。
1 资料与方法
1.1 一般资料
收集2017年1月-2019年5月在我院耳鼻喉科门诊确诊的213例感音神经性耳聋(单侧或者双侧)患儿,年龄0~14岁,男童137例,女童94例。
1.2 诊断及评估标准
入选标准:①出生时听力筛查未通过或者出生时听力筛查通过却出现迟发性听力下降;②以耳声发射、声导抗及听性脑干诱发电位、听觉稳态(≤5岁不会配合纯音测听的患儿)、以及纯音测听(>5岁且能配合患者)为检测技术,诊断为感音神经性聋(单侧或者双侧)。
排除标准:分泌性中耳炎、突发性耳聋、外中耳畸形的患者。
分级标准:根据1997年世界卫生组织耳聋分级标准[3]:大于5岁患儿以纯音测听结果为标准分级,选取500-1000-2000-4000Hz听阈的平均值。据此将听力损失进行如下分级:(0级)正常:≤25dBHL(1级)轻度:26~40dBHL;(2级)中度:41~60dB;(3级)重度:6l~80dB;(4级)极重 度:>81dB。小于5岁患儿以听性脑干诱发电位(ABR)结果为标准,ABR反应阈<35dBnHL为正常;36-50dBnHL为轻度听力下降;51-80dBnHL为中度听力下降;81-100dBnHL为重度听力下降;大于100dBnHL为极重度听力下降[4]。
1.3 研究方法
1.3.1 问卷调查
在征得聋儿家长同意下,采集受检儿童相关信息,包括:聋儿基本信息、听力状况、出生史、个人史、家族遗传性耳聋病史、母孕期情况、干预情况、辅助检查等(详见表1)。在接受基因检查前,均详细告知家长基因检测目的、内容,检测局限性、检测样本及用途、检测结果的准确性和效力以及保密条款,并签署了知情同意书。
1.3.2 专科检查
213例患儿均接受耳鼻咽喉科常规检查,排除外耳道鼓膜及中耳炎症情况,以及全身系统性检查。
1.3.3 听力学检查
1.3.3.1 纯音听阈测试
采用丹麦MADSEN Astera型纯音听力仪,在噪音<20dBSPL的隔声室中,采用降10升5测试法测试250~8000Hz气、骨导听阈阈值。选取500-1000-2000-4000HZ频率的气导听阈平均值作为主观听阈。
1.3.3.2 电反应测听检测
采用美国智听公司生产的Smart EP脑干诱发电位仪,EAR-3A气导插入式耳机。在隔音屏蔽室内,按照常规方法进行ABR检查,以Ⅴ波反应阈值作为客观听阈。
1.3.3.3 声导抗测听检测
采用丹麦国际Madsen公司的Titan中耳分析仪,EAR-3A插入式耳机,≤6个月婴儿用1KHz探测音,>6个月且<12个月小儿同时用1KHz和226Hz探测音进行检测,>12个月小儿采用226Hz探测音进行检测,以单峰A型声导抗图为正常。
1.3.3.4 耳声发射
采用GSI耳声发射仪对患儿进行初步筛查,使用插入式耳机,对患儿进行 500、1000、2000、4000HZ频率检测,4个频率均通过为正常。
1.3.4 影像学检查
对于明确诊断为感音神经性聋的患儿进行颞骨高分辨率水平+冠状位薄层CT检查,或行头颅/内听道磁共振检查。
1.3.5 耳聋基因检测
采集受检者2-3ml的外周血,使用EDTA抗凝,注入统一专用真空采血管,统一编号,并在冷冻室储藏,及时送检。具体相关流程如下:1、询问病史,一般体格检查及耳科检查、完成其他相关检查,确诊为感音神经性聋。2、完成《感音神经性聋的临床资料收集表》的填写并建立档案。3、确认是否要进行耳聋基因检测,完成《耳聋基因知情同意书》。4、予以收集患儿及父母双亲的外周血并及时送检。5、对耳聋基因结果予以解读及分析,并予以遗传咨询,提出治疗或康复建议。
1.3.6 判定标准
根据美国医学遗传学与基因组学会(ACMG)发布的变异解读指南进行致病性分析:pathogenic/致病性变异;likely pathogenic/疑似致病性变异;uncertain/临床意义未明变异;likely-benign/疑似良性变异;benign/良性变异。本检测只报告根据ACMG分级为“致病”、“疑似致病”、“临床意义未明”的变异,不报告“良性”及“疑似良性”等不具有临床意义的变异。
2 结果
2.1 耳聋情况
在213例样本中,轻度感音神经性聋为39例,占18.3%,中度感音神经性耳聋45例,占21.1%,重度感音神经性聋38例占17.8%,极重度感音神经性聋91例,占42.7%.
2.2 高危因素
统计了71份完整问卷表格中,与耳聋相关的高危因素主要是新生儿高胆红素血症,占8.5%,耳聋家族史占7%,早产儿占5.6%,宫内感染和新生儿窒息分别占2.8%,耳廓和其他的颅面部畸形和极低体重儿分别占1.4%。
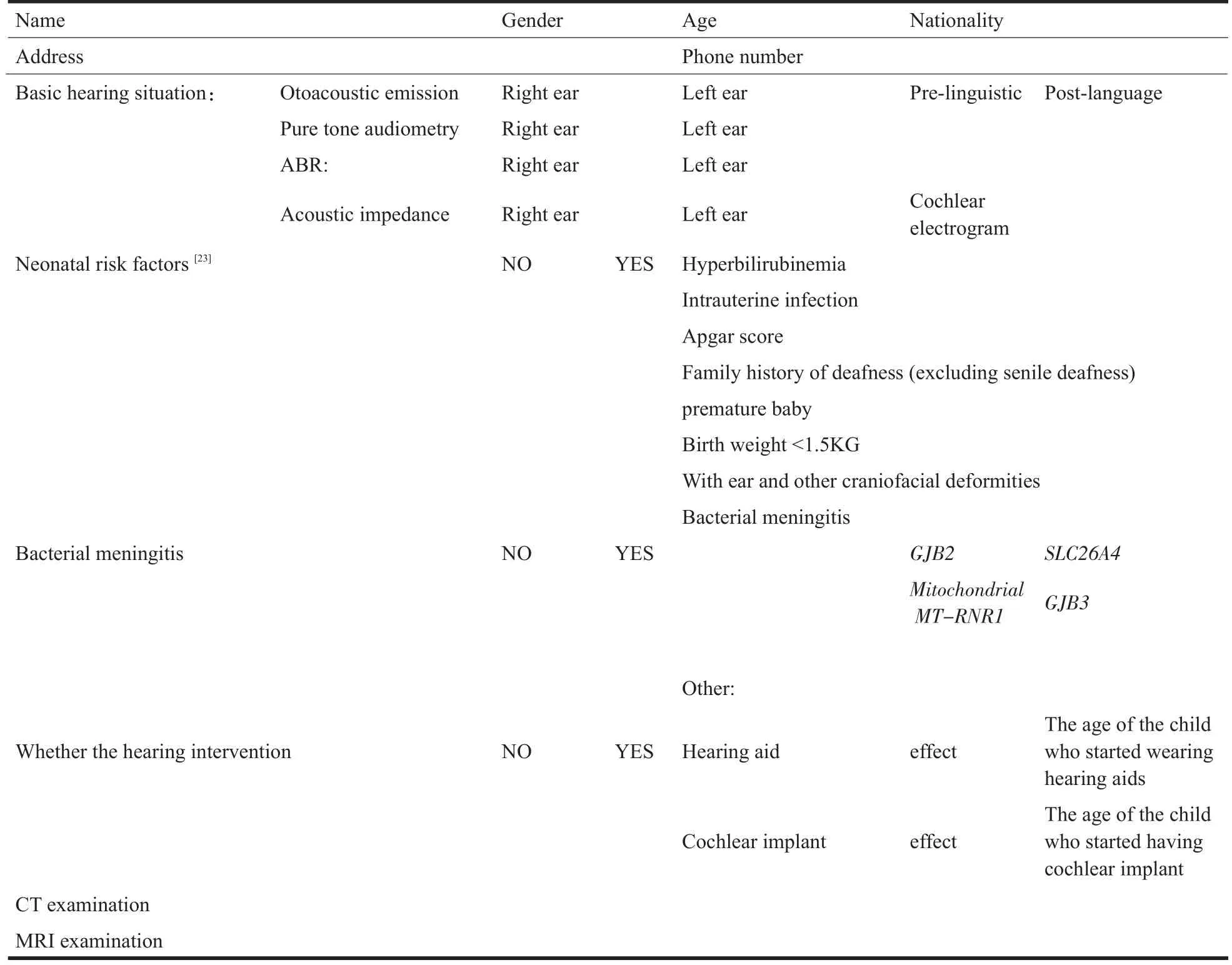
表1 感音神经性聋的临床资料收集表Table 1 Clinical data collection table for sensorineural hearing loss
2.3 内耳畸形
本研究中有172例患儿进行了高分辨率水平+冠状位薄层螺旋CT检查,以Sennaroglu[5]2010分类为标准,172例感音神经性耳聋患者高分辨率水平+冠状位薄层螺旋CT检查的结果详见表2。

表2 43例内耳畸形分布情况Table 2 Distribution of 43 cases of inner ear malformation
2.4 致病基因及突变热点
213例感音神经性聋患儿中,阳性报告56例(26.3%),疑似阳性报告52例(24.4%),未明确报告16例(7.5%),阴性报告89例(41.8%)。在阳性报告、疑似阳性报告和未明确报告中主要以GJB2、SLC26A4、USH2A、MYO15A、CDH23、MYO7A突变为主(详见表3)。

表3 124例阳性报告中各个基因的统计报告Table 3 Statistical reports of each gene in 124 positive reports
在47例GJB2基因突变样本中,以c.235delC位点突变为主的纯合突变28例,以c.235delC和c.109G>A位点复合杂合突变为主的杂合突变19例。在12例SLC26A4基因突变样本中,纯合突变6例,杂合突变6例(详见表4和表5)。

表4 阳性检测结果中的纯合突变Table 4 Homozygous mutations in positive test results

表5 阳性检测结果中的杂合突变Table 5 Hybrid mutations in positive test results
在阳性报告、疑似阳性报告和未明确报告中,108例患儿进行了听性脑干诱发电位(ABR)检查,结果显示极重度感音神经性耳聋患儿最多,有46例。GJB2基因c.235delC位点、c.109G>A位点突变和SLC26A4基因c.919-2A>G位点突变在该108例患儿中占多数(详见表6)。
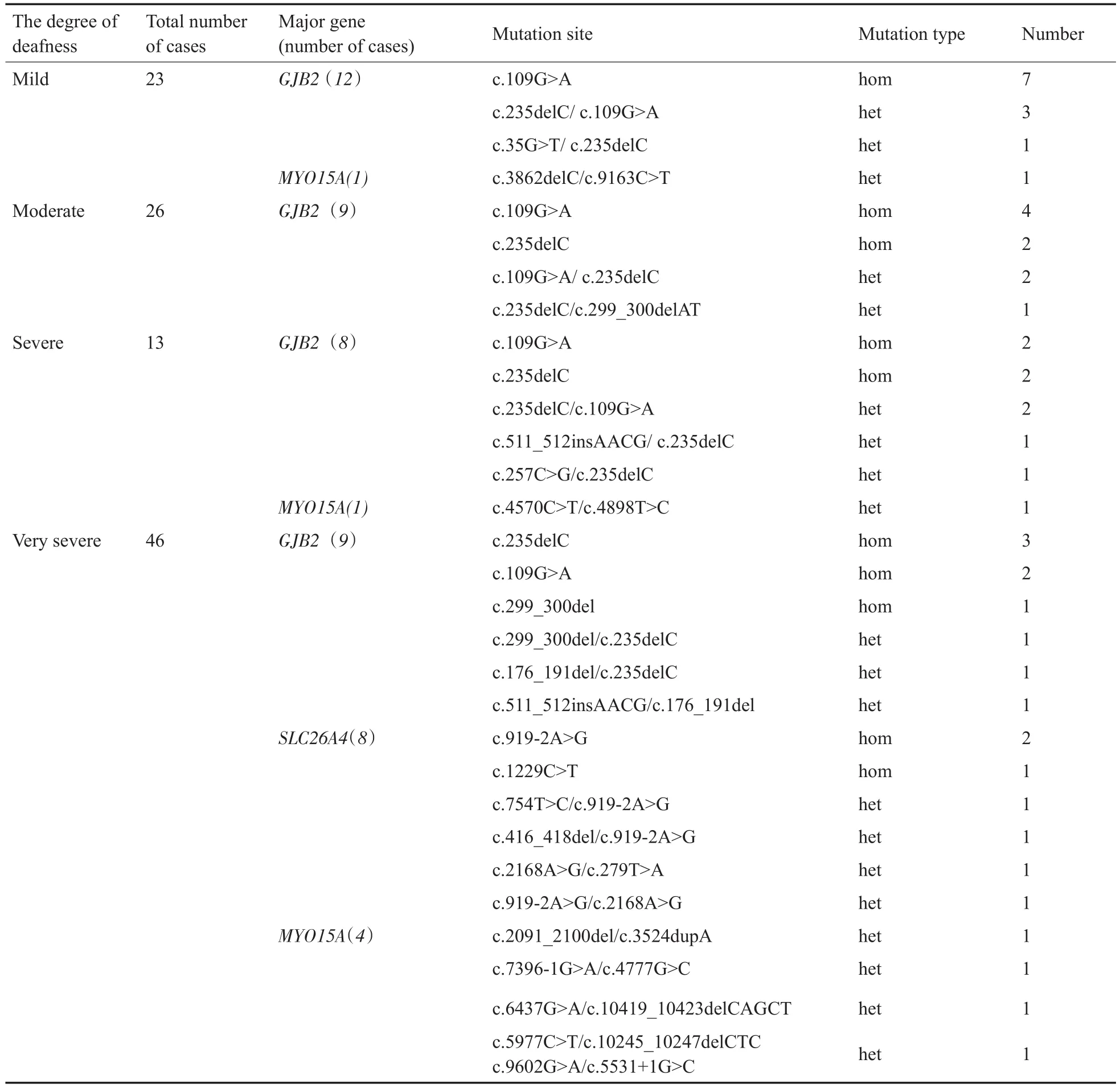
表6 在阳性报告样本中各级耳聋病例中主要的基因突变例数Table 6 Number of major gene mutations in deafness cases at all levels in positive report samples
3 讨论
在本次研究中发现重度、极重度感音神经性聋为主,占60.3%。本研究中重度、极重度感音神经性耳聋比例与国内相关文献报道的54.55%—70.43%比例相接近[6,7]。
3.1 高危因素
统计了71份完整问卷表格中,与耳聋相关的高危因素依次是新生儿高胆红素血症、耳聋家族史、早产儿、宫内感染、新生儿窒息、耳廓畸形和极低体重儿。根据杨波等人的研究显示高危因素依次为早产、新生儿高胆红素血症、NICU住院时间≥5 d[8]。由于本次研究样本量较少,无法做出相关性研究,但从结论中可以发现本次研究中得出的耳聋高危因素占比与相关研究结果相似。
3.2 内耳畸形
前庭导水管扩大是最常见的内耳畸形类型,诊断标准为高分辨率颞骨CT显示总脚至前庭水管外口的连线上前庭水管的宽度超过1.5mm[9]。本研究中最常见的畸形是单纯性前庭导水管扩大,占58.1%(25/43);其次是半规管畸形占16.3%(7/43);耳蜗畸形占16.3%(7/43)。这与李幼靖等对NSHL听力损失患者[10]的研究趋势相同。但本研究的畸形发生率较低,这可能与本研究的样本数量较少有关。本研究发现单纯性前庭导水管扩大患者经人工耳蜗植入术后,均获得良好的助听效果。另外,在本研究中发现共同腔畸形2例。此种畸形的患者术前需进行MRI和电诱发听觉脑干反应(EABR)检查,从而确认是否存在蜗神经及前庭神经,预估人工耳蜗植入术后效果。目前该2例患儿暂未行人工耳蜗手术。
3.3 耳聋基因检测技术
NGS是在Sanger测序方法的基础上得到进一步发展和创新的,通过把大量被检测的模板DNA片段放在芯片上固定,并进行杂交结合通用的DNA引物,利用不同的方法分别控制4种碱基在DNA引物上的延伸,通过检测延伸反应过程或碱基,实现DNA序列信息的检测[11]。高通量测序技术通过增加更多个体、更多基因编码区更深层次的覆盖度,来获取准确性更高的数据,已成为现阶段在基因组水平上发现致病基因的有效手段。
3.4 耳聋基因
根据数据显示[12],在欧美人群中常见的耳聋基因是 GJB2、SLC26A4、MYO15A、OTOF、CDH23 和TMC1,其中最常见的耳聋基因为GJB2。在我国,GJB2、SLC26A4、线粒体基因 mtDNA(A1555G 和C1494T突变)是最为常见的耳聋基因[13]。本次研究GJB2基因突变检出率为22.1%;SLC26A4基因突变为5.63%;USH2A基因突变为3.29%;MYO15A基因突变为2.82%;CDH23基因突变为1.41%。这与相关文献报道基本一致。USH2A、MYO15A、CDH23、MYO7A基因突变也有不同程度的检出。
3.4.1 GJB2基因
GJB2基因在普通耳聋群体中检出率约为15%~25%[14]。本研究发现47例患者存在GJB2基因单杂合或双杂合突变,检出率为22.1%。根据戴朴教授等研究结果显示,在我国GJB2基因检出率为21.01%[15],而本次研究结果与戴朴教授等研究结果相近,且略微高于戴朴教授等的研究。
本研究发现GJB2基因纯合突变28例,复合杂合突变19例。其中有8例极重度感音神经性聋的患儿行人工耳蜗植入术,术后获得良好助听效果。因此,我们认为GJB2基因突变相关的感音神经性耳聋患者行人工耳蜗植入手术后效果良好。这与相关研究[16]结果相一致。
3.4.2 SLC26A4基因
本次研究发现SLC26A4基因突变检出率仅次于GJB2,检出率为5.63%。低于戴朴等研究的SLC26A4基因突变检出率14.5%。究其原因可能是本次研究样本数量较戴朴等研究的少,检测对象重度以上耳聋比例存在差异所致。
SLC26A4基因突变与单纯前庭水管扩大和(或)内耳畸形有密切的关系[17]。在我国,约96%的前庭水管扩大的患者由SLC26A4基因突变致病。SLC26A4基因突变在人群中的检出率为20.35%(双等位基因突变19.43%,单等位基因突变0.92%),SLC26A4基因(c.919-2A>G)突变是中国大前庭水管患者群的热点突变[18]。本次研究中有12例确诊为SLC26A4基因(c.919-2A>G)突变,其中纯合突变6例,杂合突变6例,为该样本人群的热点突变。本研究中的12例SLC26A4基因突变患儿,其颞骨CT均提示前庭水管扩大,这12例患儿甲状腺无明显肿大,其中4例行人工耳蜗植入术,术后预后良好。
3.4.3 MYO15A基因
MYO15A基因突变可以导致常染色体隐性遗传非综合征型感音神经性耳聋。MYO15A编码的肌球蛋白XVa可以维持耳蜗毛细胞内肌球蛋白组织结构及毛细胞静纤毛的伸长[19]。相关研究[20]表明N-末端区域发生突变会导致肌球蛋白亚型异常,听力表型为有残余听力的非重度极重度感音神经性聋。而非N-末端 区 域(Motor、MyTH4 1、FERM1、SH3、MyTH4 2、FERM2及PDZ结构域)发生的突变,听力表型多为先天性或语前性全频重度-极重度感音神经性耳聋。在本研究中有5例MYO15A基因突变患儿为重度以上神经性耳聋,有1例为轻度感音神经性耳聋。MYO15A基因研究对感音神经性聋的诊断以及对感音神经性耳聋患儿的听力干预具有意义。
3.4.4 TMC1基因
在本次研究中发现一份TMC1基因复合杂合突变报告。该基因有2个突变位点,突变位点为c.16+2T>C/c.969C>G。c.16+2T>C导致氨基酸改变,为剪接突变。根据ACMG指南,该变异初步判定为致病性变异(Pathogenic)。c.969C>G(编码区第969号核苷酸由胞嘧啶变异为鸟嘌呤),导致氨基酸改变p.Y323X,为无义突变。根据ACMG指南,该变异初步判定为致病性变异(Pathogenic)。对于c.969C>G(编码区第969号核苷酸由胞嘧啶变异为鸟嘌呤)的突变分析已经明确[21],即导致氨基酸改变p.Y323XTCA1无义突变。但是在TMC1基因中发生了c.16+2T>C变异,剪接突变导致对蛋白产物的影响。因此,我们接下来将进行相关实验来阐明这一问题。
3.5 干预方法
在我院门诊确诊的3月龄以上的中重度感音神经性耳聋患儿,均予以配戴助听器,干预期3-6个月。重度-极重度感音神经性耳聋患儿经3-6个月助听器干预后未能得到满意的助听效果,则排除相关手术禁忌后,予以行人工耳蜗植入。绝大多数聋儿术后取得较好的助听效果,但仍有部分聋儿无明显效果,推测其原因可能与基因突变导致的病变部位有关。根据相关文献报道[22],人工耳蜗植入术后效果良好的有以下几个基因:GJB2、SLC26A4、12SrRNA(MT-RNR1:A1555G、C1494T)、OTOF、CDH23、MYO6、MYO7A、KCNQ1、TMC1、COCH、LOXHD1、MYO15A、TECTA、ACTG1、TMPRSS3;不确定的有:MYH9、POU3F4、PCDH15;植入效果差的有:CHD7、TIMM8A、DFNB59。因此,通过基因检测,推断出可能的分子病因,预测人工耳蜗植入后的效果,为遗传性聋的早期诊断和个体化治疗提供理论依据,为耳聋患者提供更精准的医学指导。
4 结论
本研究结果表明应用NGS可检测耳聋大样本群体中的相关基因突变,有助于发现以及明确新的、非热点突变位点,提高相关基因突变的检出率,同时也将为感音神经性耳聋患儿及其家属提供更全面的遗传信息。当然在临床诊疗过程中需同时关注内耳畸形及耳聋相关的新生儿高危因素,为遗传性聋的早期诊断和个体化治疗提供技术支持,为感音神经性耳聋患者提供更精准的诊疗依据,并予以及早干预。
