1 例误诊为PBC 的朗格汉斯组织细胞增生症
2020-06-23肖招英徐孟秋康子矜
肖招英,徐孟秋,康子矜
(浙江省嵊州市人民医院/ 浙大一院嵊州分院 感染科,浙江 嵊州)
0 引言
朗格汉斯组织细胞增生症(LCH),是一种罕见的组织细胞增生性疾病,以内分泌症状为首发表现的LCH 患者,主要临床表现为尿崩症、矮小症,而甲状腺受累者较少见。根据病变累及的范围,分为3 种基本类型:①多系统多灶性LCH,表现为广泛的红斑样或湿疹样皮疹,可有肝、脾、淋巴结肿大,多发性溶骨性损害及反复感染,造血系统常出现贫血、血小板减少,病情进展快。②单系统多灶性LCH,最常累及骨骼系统,病灶≥2 个,其次为淋巴结、皮肤、肺等,临床表现及预后依病灶部位及数目而异。③单灶性LCH,常发生于儿童和青少年,最常累及骨,临床表现为无性痛或痛性的溶骨性病变,偶可致病理性骨折。因为LCH 是罕见病,多见于婴幼儿或青少年,成人较少见,男性多于女性[1-2],主要累及骨骼如颅骨、脊椎、长骨、和颌骨,肺脏,肝脏,脾脏 淋巴结和皮肤[3-4]。
1 病例介绍
1.1 体格检查。患者,女,55 岁。因“乏力、尿黄”1 年余2017.12.3 入院。3 年前有 “尿崩症”,2 年前体检怀疑“肝癌”,经多家医院就诊,行肝脏增强MR、2 次肝脏穿刺活检、PET/CT 等多项检测后,肝癌依据不足,考虑M2 阴性PBC,给予“熊去氧胆酸胶囊”口服1 月后自行停药。发现“高血压”病史3年余,“糖尿病”病史6 年余,“甲状腺功能减退症”7 月余。否认服用中药史,否认毒物、放射性物质接触史。否认家族性“肝炎”、“肿瘤”等病史。患者1 年前无明显诱因下出现全身乏力不适,易疲劳,以双下肢为主,发现尿色偏黄,尿色呈逐渐加深,后呈浓茶样,偶有皮肤瘙痒,肝功能异常收入感染科住院,入院查体:体温:36.2(耳)℃,脉搏:82 次/分,呼吸:20 次/分,血压:131/86 mmHg 神清,精神一般,皮肤巩膜黄染,浅表淋巴结未及肿大,口腔牙齿有脱落,心率82 次/分,律齐,两肺未及明显啰音。腹平软,无压痛、反跳痛,肝脾肋下未及,移动性浊音阴性,双下肢无水肿。
1.2 临床诊断。初步诊断:肝功能异常原因待查:①原发性胆汁性胆管炎(PBC)?ALP、GGT 高;②肉芽肿性病变;汇管区非干酪样肉芽肿结核?无干酪样坏死、抗酸阴性;结节病?寄生虫感染?③朗格汉斯组织细胞增生症?④霍奇金淋巴瘤?近几年肝功能各项指标变化见表1,其余检查:2015-5-13 头颅CT 提示:考虑左侧基底节区、丘脑区腔隙性脑梗塞。腹部增强CT 示:肝内转移性肿瘤可能性大,右肝顶区小囊肿。胸部CT 示:两肺炎症性病变,右上肺斑点状病灶,左侧局部肋骨畸形。胃镜:①慢性浅表性胃炎伴胃窦糜烂及胃底小息肉;②贲门内缘炎症。病理示(胃窦)粘膜慢性炎伴轻度肠化。结肠镜检查提示:结肠息肉(已咬除)。病理示炎性息肉。肿瘤全套:未见异常。多次肝炎全套、EB 病毒、柯萨奇病毒、巨细胞病毒、IgG4、ANCA、抗核抗体全套均阴性。PET/CT:肝表面不光整,肝实质FDG代谢欠均匀,肝内多发稍低密度影,常规及延迟扫描未见FDG 代谢增高,建议结合病理并MR 密切随访。右侧隔心角淋巴结增大,PDG 代谢轻度增高,子宫颈部右缘斑片状FDG 代谢增高。MRCP:胰胆管未见异常改变。MRI:肝脏弥漫病变伴肝损害,炎症性病变考虑;右肝顶部小囊肿,脾大,右侧心隔角旁、肝门部及后腹膜多发淋巴结肿大。
归纳病史特点:患者为55 岁的中年女性:乏力尿黄1 年余;甲状腺功能减退症、糖尿病、肝内多发占位性病变、脾肿大、尿崩症、牙齿有脱落;2018-1-26 垂体MR:空泡蝶鞍表现;生化检查:ALP、GGT 显著升高 后期黄疸进行性升高。
两次肝脏穿刺玻片会诊后病理特点:汇管区肉芽肿形成,较多嗜酸性粒细胞浸润;部分区胆管缺失;高倍下肉芽肿内细胞核扭曲,呈“咖啡豆样”见图(1、2、3)。
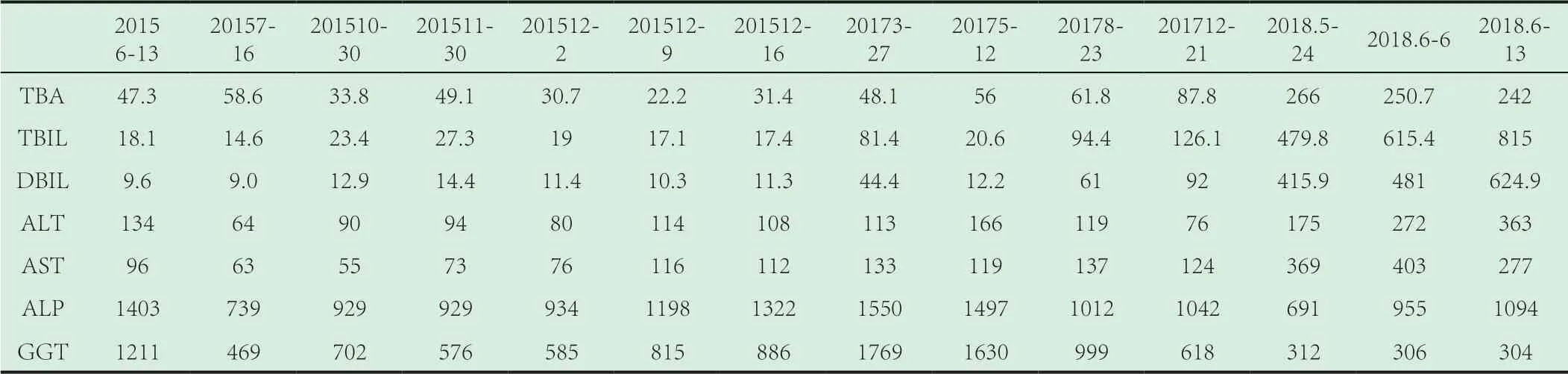
表1 近几年肝功能各项指标变化

图1 肝脏穿刺玻片会诊后病理特点:汇管区肉芽肿形成,较多嗜酸性粒细胞浸润

图2 肝脏穿刺玻片会诊后病理特点:部分区胆管缺失
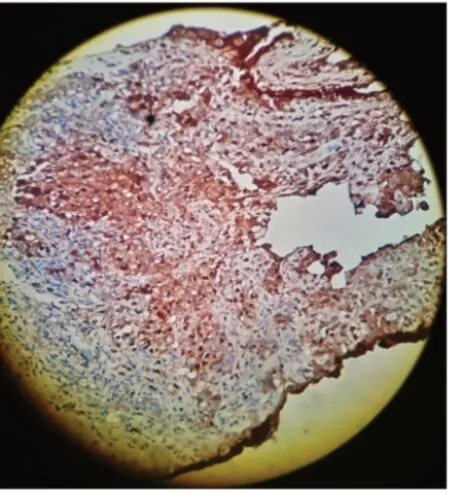
图3 肝脏穿刺玻片会诊后病理特点:高倍下肉芽肿内细胞核扭曲,呈“咖啡豆样”
2 结果
最后经临床医师及多位知名的病理科专家会诊意见确诊:朗格汉斯组织细胞增生症。明确诊断后在血液科住院,建议化疗,因多种因素未化疗,终因肝功能衰竭、肝性脑病于2018 年6 月15 在本院死亡。
3 讨论
LCH 患者肝脏增大常伴有肝功能异常,部分患者还可出现硬化性胆管炎及肝硬化,脾脏的增大可由组织细胞浸润所致,也可继发于门脉高压[5]本例特点为:①中年女性;②有糖尿病、尿崩症、甲状腺功能低下、牙齿脱落、肝脏、脾脏受损;③辅查:以肝功能受损为主,早期以r-GT AKP 反复升高,后期出现黄疸进行性上升;肝脏影像提示:肝内多发结节,垂体MR 空泡蝶鞍表现。④常规各种治疗无效;经MDT 会诊后考虑有尿崩症、甲状腺受累、糖尿病,一元论解释首先考虑LCH,再次进行阅片并加免疫组化才得以证实此病。研究显示,肝脏受累导致的黄疸和骨髓受累导致的血象抑制常有较高的病死率[6]以肝损伤为主患者预后较差,本例患者最终死于肝衰竭。与白彦报道的郎格罕组织细胞增生症五例临床分析相符[7],本例患者为多系统多病灶性类型,从误诊教训看,对于罕见病,需要感染科医师的知识面广,不能盲目相信病理结果,对出现的症状尽量以一元论解释,本例患者有两次省级病理的活检及免疫组化,均未提示此病,导致误诊,失去早期治疗的时机,后期出现黄疸进行性升高,最终死于肝功能衰竭。另一方面确诊也需要有经验的病理科专家,临床遇见少见病、尤其罕见病还需要多学科合作,才能减少误诊及漏诊。
总之,朗格汉斯组织细胞增生症极易误诊为PBC,患者具有较高的死亡率,需要临床积极积累经验对其进行及早准确诊断。
